CompositionPrincipes actifs
YESCARTA (axicabtagene ciloleucel) est une immunothérapie constituée de cellules T autologues génétiquement modifiées, dirigée contre le CD19. Pour préparer YESCARTA, les cellules T du patient sont prélevées puis génétiquement modifiées ex vivo par transduction rétrovirale pour exprimer un récepteur d'antigène chimérique (CAR) comprenant un fragment variable à chaîne simple murin (scFv) anti-CD19 relié au domaine de co-stimulation CD28 et au domaine de signalisation CD3-zêta.
Excipients
Cryostor CS10 (DMSO; dextran 40), chlorure de sodium, albumine sérique humaine (chlorure de sodium, N-acétyl-DL-tryptophane, acide caprylique, eau), 5% DMSO.
YESCARTA contient environ 300 mg de sodium par poche de perfusion.
Indications/Possibilités d’emploiYESCARTA est une immunothérapie constituée de cellules T autologues génétiquement modifiées, dirigées contre le CD19 et il est utilisé chez les patients adultes pour le traitement
·du lymphome diffus à grandes cellules B (LDGCB), ou du lymphome B de haut grade (LBHG) qui est réfractaire à la chimio-immunothérapie de première ligne ou ayant récidivé dans les 12 mois après la chimio-immunothérapie de première ligne.
·du LDGCB récidivant ou réfractaire (r/r) ou du lymphome médiastinal primitif à grandes cellules B (LMPGB) après au moins deux lignes de traitement systémique.
·du lymphome folliculaire (LF) récidivant ou réfractaire après au moins trois lignes de traitement systémique.
Posologie/Mode d’emploiLe traitement par YESCARTA doit être instauré et surveillé sous la responsabilité de médecins expérimentés dans le traitement des néoplasies hématologiques et formés pour la prise en charge de patients ayant été traités par YESCARTA, y compris pour le traitement du syndrome de relargage des cytokines (CRS) et de la neurotoxicité. La prise de YESCARTA doit avoir lieu dans un centre de traitement qualifié disposant d'un accès immédiat aux unités de soins intensifs appropriées par le personnel médical qui a été formé pour l'administration de YESCARTA. Il est indispensable de disposer d'au moins 4 doses de tocilizumab prêtes à l'emploi avant la perfusion.
YESCARTA est un médicament destiné à une perfusion unique et exclusivement destiné à une utilisation autologue et intraveineuse (voir «Mises en garde et précautions»).
La disponibilité de YESCARTA doit être confirmée avant de commencer le schéma de traitement de lymphodéplétion. La fabrication et la libération de YESCARTA nécessitent habituellement environ 3 à 4 semaines. Dans certains cas, malgré la réussite de la leucaphérèse, un patient peut ne pas pouvoir être traité par YESCARTA (voir «Propriétés/Effets»).
Prétraitement
·Un schéma de chimiothérapie lymphodéplétive consistant en l'administration de cyclophosphamide 500 mg/m2 par voie intraveineuse et de fludarabine 30 mg/m2 par voie intraveineuse doit être administré les 5e, 4e et 3e jours avant la perfusion de YESCARTA. Un nombre absolu de neutrophiles (ANC) ≥1000/μl et un nombre de plaquettes ≥75 000/μl sont recommandés avant le début de la chimiothérapie lymphodéplétive.
Évaluation clinique avant la perfusion YESCARTA
Le traitement par YESCARTA doit être retardé chez certains groupes de patients à risque (voir «Mises en garde et précautions»).
Prémédication
·Afin de réduire le risque d'une réaction à la perfusion, la prémédication avec du paracétamol par voie orale 500 à 1000 mg et de 12,5 mg à 25 mg de la diphénhydramine par voie intraveineuse ou orale, ou des médicaments équivalents, administrés environ 1 heure avant la perfusion de YESCARTA, est recommandée.
·L'utilisation de corticoïdes systémiques à titre préventif est déconseillée (voir «Interactions»).
Posologie
Une poche pour perfusion unique de YESCARTA spécifique à chaque patient avec une dispersion de cellules CAR T anti-CD19 dans env. 68 ml pour une dose cible de 2 x 106 cellules CAR T anti-CD19 par kg de poids corporel (intervalle: 1,0 x 106 – 2,0 x 106 cellules/kg), avec au maximum 2 x 108 cellules CAR T anti-CD19 pour les patients dont le poids corporel est égal ou supérieur à 100 kg.
Surveillance après la perfusion
Les signes et symptômes d'un potentiel CRS, des événements neurologiques et autres toxicités doivent être surveillés quotidiennement chez les patients pendant les 7 premiers jours suivant la perfusion dans un centre de traitement qualifié. Les médecins peuvent envisager une hospitalisation pendant les 7 premiers jours ou dès les premiers signes ou symptômes de CRS et/ou d'événements neurologiques.
Au terme des 7 premiers jours suivant la perfusion, le patient doit faire l'objet d'un suivi dont les modalités sont laissées à la discrétion du médecin.
Les patients doivent rester à proximité d'un établissement clinique qualifié (situé au maximum à 2 heures) pendant au moins 4 semaines après la perfusion.
Le patient doit également être informé sur le fait que, bien que la plupart des CRS et symptômes neurologiques surviennent dans les 4 premières semaines après la perfusion, des effets indésirables peuvent survenir à tout moment et nécessiter une assistance médicale.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Il n'existe aucune donnée suffisante concernant les patients présentant une insuffisance hépatique, permettant de tirer des conclusions pour cette population.
Patients présentant des troubles de la fonction rénale
Il n'existe aucune donnée suffisante concernant les patients présentant une insuffisance rénale, permettant de tirer des conclusions pour cette population.
Patients âgés
Les 28% de la population de l'étude étaient des patients âgés de 65 ans ou plus. L'efficacité et la sécurité étaient comparables dans tous les groupes d'âge. Sur la base de ces données, aucun ajustement posologique n'est nécessaire pour les patients âgés.
Enfants et adolescents
La sécurité et l'efficacité de YESCARTA chez les enfants et les adolescents de moins de 18 ans n'a pas été établie. Aucune donnée n'est disponible.
Mode d'administration
Administration intraveineuse.
YESCARTA est exclusivement destiné à une utilisation autologue par perfusion intraveineuse.
YESCARTA ne doit pas être irradié. Ne pas utiliser de filtre de réduction leucocytaire.
Précautions à prendre avant/pendant la manipulation ou avant/pendant l'administration du médicament
Ce médicament contient des cellules génétiquement modifiées. Les mesures habituelles de sécurité concernant la manipulation de ce type de médicament doivent être respectées. Pour les précautions particulières concernant l'élimination et remarques concernant la manipulation, voir «Remarques particulières».
YESCARTA contient des cellules sanguines humaines. Les professionnels de santé qui manipulent YESCARTA doivent donc prendre les mesures de précautions appropriées (porter des gants et des lunettes de protection), pour prévenir toute transmission potentielle de maladies infectieuses.
Préparation de YESCARTA avant l'administration
·Vérifier que l'identité (ID) du patient correspond aux identifiants du patient qui figurent sur la cassette de YESCARTA.
·La poche de perfusion de YESCARTA ne doit pas être retirée de la cassette si les informations figurant sur l'étiquette spécifique au patient ne correspondent pas au patient prévu.
·Une fois que l'ID du patient a été confirmée, la poche de perfusion de YESCARTA doit être retirée de la cassette.
·Vérifier que les informations du patient qui figurent sur l'étiquette de la cassette correspondent à l'étiquette de la poche de perfusion.
·Avant la décongélation, la poche de perfusion doit être inspectée pour détecter toute atteinte à l'intégrité de celle-ci. Si la poche de perfusion est abîmée, suivre les directives locales (ou prendre directement contact avec Gilead).
Décongélation
·La poche de perfusion doit être placée à l'intérieur d'une deuxième poche stérile ou manipulée selon les directives en vigueur localement.
·YESCARTA doit être décongelé à environ 37°C dans un bain-marie ou à sec, jusqu'à ce qu'il ne reste plus de glace visible dans la poche de perfusion. La poche de perfusion doit être mélangé délicatement pour disperser les amas de matériel cellulaire. Si des amas de cellules demeurent, continuer à mélanger délicatement la poche de perfusion. Les petits amas de matériel cellulaire doivent être dispersés en les mélangeant manuellement délicatement. YESCARTA ne doit pas être lavé, centrifugé et/ou remis en suspension dans un nouveau milieu avant la perfusion. La décongélation prend 3 à 5 minutes environ.
·Une fois décongelé, YESCARTA peut être conservé jusqu'à 3 heures à température ambiante (20°C à 25°C). Toutefois, la perfusion de YESCARTA doit commencer dans les 30 minutes suivant la fin de la décongélation.
Administration
·Ne pas utiliser de filtre de réduction leucocytaire.
·Avant la perfusion et pendant la période de surveillance, on doit disposer de tocilizumab et d'un équipement d'urgence.
·YESCARTA est uniquement destiné à un usage autologue.
·L'identité du patient doit à nouveau être vérifiée, afin de la comparer aux identifiants du patient figurant sur la poche de perfusion de YESCARTA.
·Un accès veineux central est recommandé pour l'administration de YESCARTA.
·La tubulure doit être amorcée avec une solution de chlorure de sodium stérile 9 mg/mL (0.9%) (0.154 mmol de sodium par mL) pour l'injection, avant la perfusion.
·La totalité du contenu de la poche de perfusion de YESCARTA doit être perfusée en 30 minutes, par gravité ou au moyen d'une pompe péristaltique.
·La poche de perfusion doit être délicatement agitée pendant la perfusion de YESCARTA pour éviter l'agglutination des cellules.
·Une fois que la totalité du contenu de la poche de perfusion a été perfusée, la poche de perfusion et la tubulure doivent être rincées par rétro-amorçage avec 10 a 30 mL de solution de chlorure de sodium 9 mg/mL (0.9%) en gardant le même débit de perfusion afin de s'assurer que la totalité de dose de YESCARTA a été administrée.
Contre-indicationsHypersensibilité au principe actif, à l'un des excipients (voir «Composition») ou à l'une des substances figurant dans les contre-indications de l'information professionnelle de la fludarabine ou du cyclophosphamide.
Mises en garde et précautionsGénéralités
Les mises en garde et précautions liées à la chimiothérapie lymphodéplétive doivent être prises en compte.
Motifs entraînant un report du traitement
En raison des risques associés au traitement par YESCARTA, la perfusion doit être reportée si un patient présente l'une des conditions suivantes:
·Effets indésirables sévères non résolus (en particulier réactions pulmonaires, réactions cardiaques ou hypotension), y compris les effets dus à des chimiothérapies précédentes.
·Inflammation active ou infection non contrôlée.
·Graft-versus host disease (GvHD) active.
·Développement d'une aggravation clinique importante du lymphome entraînant un dysfonctionnement organique médicalement significatif ou une aggravation clinique après une chimiothérapie lymphodéplétive.
Dans certains cas, le traitement par YESCARTA peut être reporté après l'application du schéma de chimiothérapie lymphodéplétive. Si la perfusion est retardée de plus de 2 semaines après que le patient a reçu la chimiothérapie lymphodéplétive, il convient d'appliquer de nouveau le schéma de chimiothérapie lymphodéplétive (voir «Posologie/Mode d'emploi»).
Les patients traités par YESCARTA ne devraient pas faire de don de sang, d'organes, de tissus et de cellules pour une greffe.
YESCARTA est exclusivement destiné à une utilisation autologue et ne doit en aucun cas être administré à d'autres patients. Avant la perfusion, l'identité du patient doit correspondre aux identifiants du patient figurant sur la poche de perfusion et la cassette de YESCARTA. Ne perfusez pas YESCARTA, si les informations figurant sur l'étiquette spécifique au patient ne correspondent pas au patient prévu.
Réactions d'hypersensibilité
Des réactions graves et potentiellement mortelles liées à la perfusion, telles que l'anaphylaxie (y compris le choc anaphylactique, l'arrêt cardiaque et l'arrêt respiratoire) ont été rapportées avec YESCARTA. Des réactions allergiques peuvent se produire avec la perfusion de YESCARTA.
Des réactions d'hypersensibilité graves, y compris une anaphylaxie, peuvent être dues au DMSO ou à la gentamicine résiduelle présente dans YESCARTA.
Localisation cérébrale d'un lymphome, lymphome du système nerveux central et localisation cardiaque d'un lymphome
Il n'existe aucune expérience clinique relative à l'utilisation de YESCARTA chez les patients ayant des antécédents ou présentant un lymphome primaire du SNC en phase aiguë, chez les patients présentant des cellules malignes dans le liquide cérébrospinal, chez les patients atteints de métastases cérébrales, ou chez les patients présentant une localisation cardiaque d'un lymphome.
Maladies concomitantes
Les patients atteints d'une maladie du système nerveux central (SNC) active ou présentant des troubles préexistants des fonctions organiques, notamment une fonction pulmonaire, cardiaque, rénale ou hépatique amoindrie en raison de maladies préexistantes, ainsi que les patients atteints de thrombocytopénie ou présentant un faible taux de fibrinogène sont probablement susceptibles d'être plus vulnérables aux conséquences des effets indésirables décrits ci-dessous et nécessitent une attention particulière. En outre, il n'existe aucune expérience clinique relative à l'utilisation de YESCARTA chez les patients présentant une dysfonction modérée ou sévère des fonctions organiques.
Syndrome de relargage des cytokines
Presque tous les patients ont présenté, à des degrés divers, un CRS. Un CRS sévère, y compris mettant en jeu le pronostic vital et des réactions fatales ont été très fréquemment observés avec YESCARTA, et le délai d'apparition du syndrome se situait entre 1 et 12 jours dans ZUMA-1 et ZUMA-7 et entre 1 et 15 jours dans ZUMA-5 (voir «Effets indésirables»).
Le diagnostic de CRS requiert d'exclure les autres causes possibles d'une réaction inflammatoire systémique, y compris une infection.
Prise en charge du syndrome de relargage des cytokines associé à YESCARTA
Assurez-vous qu'au moins 4 doses de tocilizumab (un inhibiteur du récepteur de l'interleukine-6 (IL-6)), sont disponibles pour chaque patient avant la perfusion de YESCARTA.
Des algorithmes de traitements ont été développés, pour atténuer certains symptômes de CRS ressentis par les patients traités par YESCARTA. Cela inclut l'utilisation de tocilizumab ou de tocilizumab associé à des corticoïdes, pour un CRS modéré, sévère ou mettant en jeu le pronostic vital (voir résumé dans le Tableau 1 ci-dessous). Les patients présentant un CRS de Grade 2 ou supérieur (p.ex. une hypotension ne répondant pas à un remplissage liquidien, ou une hypoxie nécessitant une supplémentation en oxygène) doivent être surveillés par monitoring du rythme cardiaque par télémétrie et oxymétrie de pouls en continu. Pour les patients présentant un CRS sévère, envisager de réaliser une échocardiographie afin d'évaluer la fonction cardiaque. En cas de CRS sévère ou mettant en jeu le pronostic vital, envisager un traitement médical symptomatique intensif.
YESCARTA ne doit pas être administré à des patients atteints d'infections actives ou de maladies inflammatoires tant que ces maladies ne sont pas sous contrôle.
Le CRS est notamment associé à des défaillances d'organes (par exemple foie, rein, cœur et poumon). En outre, une aggravation des pathologies organiques sous-jacentes peut survenir dans le cadre d'un CRS. Un taux de fibrinogène abaissé peut, en particulier dans un contexte de thrombocytopénie, accroître le risque d'hémorragies. Les patients présentant des troubles médicalement significatifs de la fonction cardiaque doivent être traités selon les standards de médecine intensive et des mesures telles qu'une échocardiographie doivent être envisagées.
Il convient de chercher une lymphohistiocytose hémophagocytaire/un syndrome d'activation des macrophages (HLH/MAS) chez les patients atteints de CRS sévère ou d'un CRS ne répondant pas au traitement.
Tableau 1: Détermination du grade et recommandations de prise en charge du CRS
|
Grade du CRSa
|
Tocilizumab
|
Stéroïdes
| |
Grade 1
Symptômes nécessitant uniquement un traitement symptomatique (p.ex. fièvre, nausées, fatigue, céphalées, myalgies, malaise).
|
Si aucune amélioration après 24 heures, administrer le tocilizumab 8 mg/kg par voie intraveineuse sur 1 heure (maximum 800 mg).
|
n. d./n. a.
| |
Grade 2
Symptômes nécessitants et répondants à une intervention modérée.
Besoin en oxygène FiO2 < 40% ou hypotension répondant à un remplissage vasculaire ou à un vasopresseur à faible dose ou toxicité d'organe de Grade 2b.
|
Administrer le tocilizumabc 8 mg/kg par voie intraveineuse sur 1 heure (maximum 800 mg).
Répéter l'administration du tocilizumab toutes les 8 heures si nécessaire en cas d'absence de réponse à un remplissage vasculaire ou à l'augmentation de l'oxygénothérapie. Se limiter à un maximal de 3 doses par 24 heures; un total de 4 doses maximum en cas d'absence d'amélioration clinique des signes et des symptômes de CRS.
|
Prendre en charge comme un Grade 3 si aucune amélioration dans les 24 heures suivant le début du traitement par tocilizumab.
| |
Grade 3
Symptômes nécessitants et répondants à une intervention agressive.
Besoin en oxygène FiO2 ≥40% ou hypotension nécessitant un vasopresseur à haute dose ou de multiples vasopresseurs ou toxicité d'organe de Grade 3 ou élévation des transaminases de Grade 4.
|
Identique au Grade 2.
|
Administrer 1 mg/kg de méthylprednisolone par voie intraveineuse deux fois par jour ou une dose équivalente de dexaméthasone (par ex. 10 mg par voie intraveineuse toutes les 6 heures).
Continuer les corticoïdes jusqu'à ce que l'effet soit de Grade ≤1, puis diminuer progressivement sur 3 jours. En l'absence d'amélioration, la prise en charge est identique au Grade 4 (voir ci-dessous).
| |
Grade 4
Symptômes mettant en jeu le pronostic vital.
Besoin d'une assistance respiratoire ou d'une hémodialyse veinoveineuse continue (CVVHD) ou défaillance d'organe de Grade 4 (à l'exclusion de l'élévation des transaminases).
|
Identique au Grade 2.
|
Administrer 1000 mg/jour de méthylprednisolone par voie intraveineuse pendant 3 jours; en cas d'amélioration, prendre en charge comme indiqué cidessus.
Si aucune amélioration, envisager 1000 mg de méthylprednisolone par voie intraveineuse deux à trois fois par jour ou un autre traitement.d.
|
a Lee et al 2014
b Voir le Tableau 2 pour le traitement des effets indésirables neurologiques
c Voir l'information professionnelle du tocilizumab pour de plus amples informations
d L'instauration de l'autre traitement comprend (mais sans s'y limiter): anakinra, siltuximab, ruxolitinib, cyclophosphamide, immunoglobuline intraveineuse (IgIV) et globuline anti-thymocytes (ATG)
Effets indésirables neurologiques
Des effets indésirables neurologiques sévères, également appelés syndrome de neurotoxicité lié aux cellules effectrices de l'immunité (ICANS), pouvant mettre en jeu le pronostic vital ou être fatals ont été très fréquemment observés chez les patients traités par YESCARTA (voir «Effets indésirables»). La durée médiane jusqu'à la survenue de ces effets était de 6 jours (intervalle: 1 à 133 jours) dans ZUMA-1 et ZUMA-7 et de 7 jours (intervalle: 1 à 177 jours) dans ZUMA-5. Des cas d'état d'épilepsie ont été observés pendant la phase post-marketing. Chez les patients ayant des antécédents de troubles du système nerveux central (SNC), tels que des convulsions ou une ischémie vasculaire cérébrale, le risque pourrait être accru. Des cas graves, voire fatals, d'œdème cérébral ont été rapportés chez des patients traités par YESCARTA. Les patients doivent être surveillés pour détecter d'éventuels signes et symptômes d'effets indésirables neurologiques/d'ICANS (Tableau 2).
Les patients présentant des toxicités neurologiques/d'ICANS de Grade 2 ou supérieur doivent être surveillés en continu par monitoring du rythme cardiaque par télémétrie et oxymétrie de pouls. Administrer un traitement symptomatique médical intensif en cas de toxicités neurologiques/d'ICANS sévères ou mettant en jeu le pronostic vital.
Le lévétiracétam peut être envisagé pour la prévention des convulsions associées à des effets indésirables neurologiques. Des algorithmes de traitements ont été développés pour prendre en charge les patients traités par YESCARTA afin d'atténuer les effets indésirables neurologiques. Ils incluent l'utilisation de tocilizumab (en cas de présence associée d'un CRS) et/ou de corticoïdes, pour des effets indésirables neurologiques légers, modérés, sévères ou mettant en jeu le pronostic vital (voir résumé dans le Tableau 2 ci-dessous).
Tableau 2: Détermination du grade et recommandations de prise en charge des effets indésirables neurologiques/d'ICANS
|
Evaluation du grade a
|
CRS associé
|
Pas de CRS associé
| |
Grade 2
|
Administrer le tocilizumab selon les recommandations présentées dans le Tableau 1 pour la prise en charge du CRS de Grade 2.
En l'absence d'amélioration dans les 24 heures après le début du traitement par tocilizumab, administrer 10 mg de dexaméthasone par voie intraveineuse toutes les 6 heures si le patient ne reçoit pas déjà un autre corticoïde. Continuer la dexaméthasone jusqu'à ce que l'événement soit de Grade 1 ou inférieur, puis diminuer progressivement sur 3 jours.
|
Administrer 10 mg de dexaméthasone toutes les 6 heures par voie intraveineuse.
Continuer la dexaméthasone jusqu'à ce que l'événement soit de Grade 1 ou inférieur, puis diminuer progressivement sur 3 jours.
| |
Envisager l'administration de lévétiracétam pour la prévention des convulsions.
| |
Grade 3
|
Administrer le tocilizumab selon les recommandations présentées dans le Tableau 1 pour la prise en charge du CRS de Grade 2.
De plus, administrer 10 mg de dexaméthasone par voie intraveineuse avec la première dose de tocilizumab et répéter l'administration toutes les 6 heures. Continuer la dexaméthasone jusqu'à ce que l'événement soit de Grade 1 ou inférieur, puis diminuer progressivement sur 3 jours.
|
Administrer 10 mg de dexaméthasone toutes les 6 heures par voie intraveineuse.
Continuer la dexaméthasone jusqu'à ce que l'événement soit de Grade 1 ou inférieur, puis diminuer progressivement sur 3 jours.
| |
Envisager l'administration de lévétiracétam pour la prévention des convulsions.
| |
Grade 4
|
Administrer le tocilizumab selon les recommandations présentées dans le Tableau 1 pour la prise en charge du CRS de Grade 2. Administrer de la méthylprednisolone 1000 mg par voie intraveineuse avec la première dose de tocilizumab et continuer la méthylprednisolone 1000 mg par jour par voie intraveineuse pendant 2 jours de plus; en cas d'amélioration, prendre en charge comme indiqué ci-dessus.
|
Administrer 1000 mg/jour de méthylprednisolone par voie intraveineuse pendant 3 jours; en cas d'amélioration, prendre en charge comme indiqué ci-dessus.
En l'absence d'amélioration, envisager 1000 mg de méthylprednisolone par voie intraveineuse deux à trois fois par jour ou un traitement alternatifb.
| |
Envisager l'administration de lévétiracétam pour la prévention des convulsions.
|
a Sévérité basée sur les Critères de terminologie communs pour les événements indésirables
b L'instauration de l'autre traitement comprend (mais sans s'y limiter): anakinra, siltuximab, ruxolitinib, cyclophosphamide, IgIV et ATG
Infections et neutropénies fébriles
Des infections sévères ont très fréquemment été observées en lien avec YESCARTA (voir «Effets indésirables»). Chez des patients immunodéprimés, des infections opportunistes mettant en jeu le pronostic vital et à évolution létale, y compris des infections fongiques disséminées, ont été rapportées.
Les patients doivent faire l'objet d'une surveillance pour détecter l'apparition de signes et de symptômes d'infection pendant et après la perfusion de YESCARTA et traités en conséquence. Un principe actif antibactérien doit être administré de manière préventive selon les recommandations habituelles locales. Une neutropénie fébrile a été observée chez des patients après la perfusion de YESCARTA et peut être concomitante avec un CRS. En cas de neutropénie fébrile, rechercher une infection et prendre en charge avec des antibiotiques à large spectre, des liquides et d'autres traitements médicaux de soutien appropriés. Les patients présentant un taux de protéine C-réactive (PCR) s'élevant à > 100 mg/l ont été exclus des essais cliniques.
Chez des patients immunosupprimés, y compris des patients traités par YESCARTA, des cas d'infections opportunistes à évolution létale et mettant en jeu le pronostic vital, y compris des infections fongiques disséminées et une réactivation virale (p.ex. VHH-6 et leucoencéphalopathie multifocale progressive) ont été rapportés. Chez les patients présentant des évènements neurologiques, il convient de prendre en compte de telles infections et de procéder aux examens diagnostiques adéquats.
Réactivation virale
Une réactivation du virus de l'hépatite B (VHB), entraînant dans certains cas une hépatite fulminante, une insuffisance hépatique et le décès, peut survenir chez les patients traités par des médicaments dirigés contre les cellules B. Les patients présentant une infection par le VHB, le VHC ou le VIH ont été exclus des études cliniques, on ne dispose donc d'aucune donnée issue d'études cliniques chez ces patients. Un dépistage sérologique du VHB, VHC et du VIH doit être réalisé avant de récolter les cellules pour fabriquer le produit, conformément aux recommandations cliniques.
Chez les patients dont l'anamnèse comporte une infection par le VHB ou le VHC ou chez les patients qui ont été traités contre le VHB ou le VHC, le virus doit être non détectable par un test sensible de détection convenable, avant le traitement par YESCARTA.
D'autres cas de réactivation du virus HHV-6 mettant en jeu le pronostic vital et à évolution létale ont été rapportés.
Cytopénies prolongées
Les patients peuvent présenter des cytopénies se prolongeant pendant plusieurs semaines et plusieurs mois après la chimiothérapie lymphodéplétive et la perfusion de YESCARTA et ils doivent être traités conformément aux recommandations habituelles. Des cytopénies prolongées de Grade 3 ou supérieur, y compris thrombopénie, neutropénie et anémie, ont très fréquemment été observées après la perfusion de YESCARTA. L'hémogramme du patient doit être surveillé après la perfusion de YESCARTA.
Hypogammaglobulinémie
Comme YESCARTA contient un récepteur antigénique chimérique anti-CD19, une aplasie des cellules B peut survenir chez les patients traités par YESCARTA, ce qui conduit à une hypogammaglobulinémie (voir «Effets indésirables»). Une hypogammaglobulinémie rend les patients plus vulnerables aux infections. Une hypogammaglobulinémie a été très fréquemment observée chez les patients traités par YESCARTA. Surveillez les taux d'immunoglobulines après le traitement par YESCARTA et prenez des mesures de prévention des infections, prophylaxie antibiotique et traitement substitutif par immunoglobulines conformément aux recommandations habituelles en cas d'infections récurrentes.
Vaccins vivants
La sécurité d'une immunisation par des vaccins viraux vivants pendant ou après le traitement par YESCARTA n'a pas été étudiée. La vaccination avec des vaccins viraux vivants n'est pas recommandée pendant au moins 6 semaines avant le début de la chimiothérapie lymphodéplétive, pendant le traitement par YESCARTA et jusqu'à la restauration du système immunitaire après le traitement par YESCARTA.
Tumeurs malignes secondaires
Des tumeurs malignes secondaires peuvent se développer chez les patients traités par YESCARTA.
Des tumeurs malignes à cellules T ont été rapportées après le traitement de tumeurs malignes hématologiques par une immunothérapie constituée de cellules T autologues génétiquement modifiées, ciblant BCMA et CD19, y compris le traitement par YESCARTA. Des tumeurs malignes à cellules T matures, y compris des tumeurs CAR-positives, peuvent survenir en l'espace de quelques semaines à quelques mois après la perfusion et être fatales (voir «Effets indésirables»).
Les tumeurs malignes secondaires, notamment les tumeurs malignes secondaires d'origine hématologique, doivent être recherchées tout au long de la vie. Si une tumeur maligne secondaire d'origine hématologique est détectée, contacter le laboratoire pharmaceutique afin d'obtenir des instructions pour le prélèvement d'échantillons du patient à des fins d'analyse.
Syndrome de lyse tumorale (SLT)
Un SLT, pouvant être sévère, a été occasionnellement observé. Afin de minimiser le risque de SLT, les patients dont le taux d'acide urique est élevé ou dont la charge tumorale est élevée doivent recevoir de l'allopurinol, ou une prophylaxie alternative, avant la perfusion de YESCARTA. Les signes et les symptômes du SLT doivent être surveillés et les événements doivent être gérés conformément aux recommandations habituelles.
Traitement antérieur par anti-CD19
L'expérience de YESCARTA chez les patients exposés à un traitement préalable dirigé contre le CD19 est limitée. YESCARTA n'est pas recommandé chez les patients ayant présenté une rechute avec une maladie CD19-négative suite à un traitement antérieur par anti-CD19.
Excipients
YESCARTA contient environ 300 mg de sodium par perfusion, ce qui équivaut à 15% de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte.
InteractionsAucune étude pharmacocinétique ou pharmacodynamique n'a été réalisée pour l'enregistrement d'interactions avec YESCARTA.
L'utilisation prophylactique de corticoïdes systémiques peut altérer l'activité de YESCARTA et n'est donc pas recommandée avant la perfusion (voir «Posologie/Mode d'emploi»).
L'administration de corticoïdes conformément aux recommandations de traitement des toxicités n'a aucun effet sur la propagation et la persistance des cellules CAR-T.
Grossesse, allaitementFemmes en âge de procréer/Contraception chez l'homme et la femme
Sur la base de l'information professionnelle de la fludarabine et du cyclophosphamide, les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant la préparation à la chimiothérapie ainsi qu'au moins 6 mois après la perfusion de YESCARTA. Vous trouverez des informations concernant la nécessité d'utiliser une méthode de contraception efficace pour les patientes qui reçoivent une chimiothérapie lymphodéplétive dans les informations professionnelles de la fludarabine et du cyclophosphamide.
Il n'existe aucune donnée suffisante concernant l'exposition pour établir des recommandations d'utilisation d'une contraception au-delà de 6 mois après le traitement par YESCARTA.
Il faut enjoindre les hommes à utiliser une méthode de contraception efficace pendant la préparation de la chimiothérapie ainsi qu'au moins 6 mois après la perfusion de YESCARTA.
Grossesse
Aucune étude n'a été effectuée chez l'animal avec YESCARTA, afin de déterminer si la prise chez une femme enceinte est susceptible de porter atteinte au fœtus (voir «Données précliniques»).
Le statut de grossesse des femmes en âge de procréer doit être communiqué avant le début du traitement par YESCARTA.
On ignore si YESCARTA peut potentiellement être transmis au fœtus. Sur la base du mécanisme d'action, si elles passent la barrière placentaire, les cellules transduites peuvent avoir un effet toxique sur le fœtus, y compris une lymphopénie en cellules B. Par conséquent, YESCARTA n'est pas recommandé chez les femmes enceintes et toute grossesse après perfusion de YESCARTA doit être discutée avec le médecin traitant.
Allaitement
On ignore si YESCARTA passe dans le lait maternel. Les femmes qui allaitent doivent être informées du risque potentiel pour l'enfant allaité.
Fertilité
Aucune donnée clinique n'est disponible sur l'effet de YESCARTA sur la fertilité. Les effets sur la fertilité masculine et féminine n'ont pas été évalués dans des études effectuées chez l'animal.
Effet sur l’aptitude à la conduite et l’utilisation de machinesYESCARTA a une influence modérée sur l'aptitude à la conduite ou à l'utilisation des machines. Après la perfusion de YESCARTA, il faut s'abstenir de conduire des véhicules ou d'utiliser des machines lourdes ou potentiellement dangereuses pendant au moins 8 semaines ou jusqu'à la disparition des effets indésirables neurologiques.
Effets indésirablesRésumé du profil de sécurité
Les données de sécurité décrites dans cette section proviennent d'un total de 402 patients adultes qui ont été traités par YESCARTA dans le cadre de trois études cliniques pivots multicentriques (ZUMA-1, ZUMA-7 et ZUMA-5), dont 214 patients atteints de LDGCB, 8 patients atteints de LMPGB, 28 patients atteints de LBHG et 124 patients atteints de LF, et en outre 28 patients chez lesquels la classification était absente, n'avait pas été confirmée ou d'autres informations avaient été fournies, ainsi que de données rapportées après commercialisation.
Expériences issues des études cliniques ZUMA-1, ZUMA-7 et ZUMA-5
Lymphome diffus à grandes cellules B et lymphome médiastinal primitif à grandes cellules B
Dans une étude à un bras, les patients atteints de lymphome B non Hodgkinien à cellules B agressif récidivant ou réfractaire ont été traités par YESCARTA. Sept patients ont été traités en phase 1 et 101 patients en phase 2 (N = 108).
Les données de sécurité provenant de l'étude ZUMA-1 reflètent l'exposition à une dose unique de YESCARTA dans une étude de phase 1/2, dans laquelle 108 patients atteints de LDGCB ou LMPGB récidivant ou réfractaire ont été traités par YESCARTA après au moins deux lignes de traitement systémique. Les données décrites proviennent de l'analyse du suivi à 54 mois, dans laquelle la durée médiane réelle du suivi était de 23.5 mois (intervalle: 0,3 à 67,8 mois).
Les effets indésirables les plus significatifs et les plus fréquents sont le CRS (93%), les encéphalopathies (60%) et les infections (40%). Des neutropénies, thrombopénies et anémies de Grade 3 ou supérieur, et toujours présentes au jour 30 ou plus, sont survenues chez 27%, 23% et 10% des patients respectivement.
Des effets indésirables sévères sont survenus chez 51% des patients. Les effets indésirables sévères les plus fréquents (≥5%) sont, entre autres l'encéphalopathie (22%), des infections par agent pathogène non spécifié (15%), des infections bactériennes (6%), des infections virales (6%), une neutropénie fébrile (5%) et de la fièvre (5%).
Les effets indésirables non hématologiques de Grade 3 ou supérieur les plus fréquents (≥5%) sont, entre autres, l'encéphalopathie (31%), des infections par agent pathogène non spécifié (19%), un CRS (11%), des infections bactériennes (9%), des infections virales (6%), un délire (6%), une hypotension (6%), une augmentation des transaminases (6%) et une hypertension (6%).
Lymphome diffus à grandes cellules B et lymphome B de haut grade
Les données de sécurité provenant de l'étude ZUMA-7 reflètent l'exposition à une dose unique de YESCARTA dans une étude de phase 3, dans laquelle 170 patients atteints principalement de LDGCB ou LBHG, récidivants ou réfractaires après une première ligne de traitement, ont été traités par YESCARTA. Les données décrites ci-dessous proviennent de l'analyse principale, avec une durée du suivi de 19,6 mois (intervalle: 1,5 à 36,9 mois).
Les effets indésirables les plus importants et les plus fréquents étaient le CRS (92%), les encéphalopathies (49%) et les infections (44%). Une neutropénie, thrombopénie et anémie de grade 3 ou supérieur, encore présentes au jour 30 ou ultérieurement, sont survenues chez, respectivement, 26%, 6% et 3% des patients.
Des effets indésirables graves sont survenus chez 51% des patients. Les effets indésirables graves les plus fréquents (≥5%) étaient entre autres un CRS (17 %), une encéphalopathie (16%), des infections par des agents pathogènes non spécifiés (7%) et de la fièvre (6%).
Les effets indésirables non hématologiques de grade 3 ou supérieur les plus fréquents (≥5%) étaient entre autres une encéphalopathie (19%), des infections par des agents pathogènes non spécifiés (8%), un CRS (6%) et des infections bactériennes (5%).
Dans l'étude ZUMA-7, les patients ont reçu après randomisation soit une perfusion unique de YESCARTA, soit le traitement standard (défini comme une chimio-immunothérapie standard à 2 ou 3 cycles [R-ICE, R-DHAP, R-DHAX, R-ESHAP ou R-GDP] suivie d'un traitement à haute dose (THD) et d'une greffe de cellules souches [GCS] autologue chez des patients dont la maladie répondait au traitement). Une sélection d'effets indésirables identifiés en relation avec YESCARTA en comparaison avec le traitement standard, est présentée dans le tableau 3.
Lymphome folliculaire
Les données de sécurité décrites dans cette section reflètent l'exposition à YESCARTA dans ZUMA-5, une étude de phase 2 dans laquelle 124 patients atteints de LF récidivant//réfractaire ont reçu des cellules CAR T-positives, sur la base d'une dose recommandée en fonction du poids. Les données décrites proviennent d'une analyse de suivi à 24 mois dans laquelle la durée médiane de suivi réel était de 26.6 mois (intervalle: 0,3 à 44,3 mois).
Les effets indésirables les plus significatifs et les plus fréquents sont le CRS (78%), les infections (59%) et les encéphalopathies (47%). Des neutropénies, thrombopénies et des anémies de Grade 3 ou supérieur, encore présentes au jour 30 ou plus, sont survenues chez 27%, 10% et 7% des patients respectivement.
Des effets indésirables sévères sont survenus chez 48% des patients. Les effets indésirables sévères les plus fréquents incluaient: encéphalopathie (19%), infections par des agents pathogènes non spécifiés (14%), CRS (6%), fièvre (5%), infections bactériennes (5%), infections virales (5%), neutropénie fébrile (3%) et neutropénie (2%).
Les effets indésirables de Grade 3 ou supérieur les plus fréquents incluaient: lymphocytopénie (99%), leucopénie (94%), neutropénie (92%), thrombopénie (35%), anémie (35%), hypophosphatémie (23%) et hyponatrémie (9%).
Les effets indésirables non hématologiques de Grade 3 ou supérieur les plus fréquents incluaient: encéphalopathie (14%), infections par des agents pathogènes non spécifiés (12%), CRS (6%), hypertension (4%), délire (4%), infections virales (4%), et thrombose (4%).
Résumé des effets indésirables
Les effets indésirables décrits dans cette section ont été identifiés chez des patients exposés à YESCARTA, atteints de LDGCB ou de LMPGB dans ZUMA-1 (n=108), de LDGCB ou LBHG dans ZUMA-7 (n=170) et atteints de LF dans ZUMA-5 (n=124) ainsi que dans les rapports post marketing. Ces effets indésirables sont présentés par classe de systèmes d'organes et par fréquence. Les fréquences sont définies comme suit: très fréquent (≥1/10); fréquent (≥1/100, < 1/10); occasionnel (≥1/1000, < 1/100); rare (≥1/10'000, < 1000); très rare (< 1/10'000). Dans chaque groupe de fréquence, les effets indésirables sont présentés dans un ordre de gravité décroissant.
Infections et infestations:
Très fréquent: infections par des agents pathogènes non spécifiés (32%), infections virales (19%), infections bactériennes (12%).
Fréquent: infections fongiques.
Affections hématologiques et du système lymphatique:
Très fréquent: neutropénie fébrile (10%), neutropénie (93%)*, lymphopénie (99%)*, leucopénie (95%)*, anémie (45%)*, thrombopénie (37%)*.
Fréquent: coagulopathiea.
Affections du système immunitaire:
Très fréquent: CRS (88%), immunoglobulines diminuées (15%)b.
Fréquent: réactions liées à la perfusion.
Occasionnel: hypersensibilité, histiocytose hémophagocytaire.
Néoformations:
Rare: tumeurs malignes à cellules T.
Troubles du métabolisme et de la nutrition:
Très fréquent: hyponatrémie (14%)*, hypophosphatémie (23%)*, hyperuricémie (14%)*, #, appétit diminué (27%)c.
Fréquent: hyperglycémie*, hypokaliémie*, hypocalcémie*, hypoalbuminémie*, déshydratationd, perte de poids.
Affections psychiatriques:
Très fréquent: délire (13%)e, insomnie (13%),
Fréquent: Anxiété, troubles affectifsf.
Affections du système nerveux:
Très fréquent: encéphalopathie (51%)g, tremblements (28%)h, céphalées (33%)i, sensation vertigineuse (22%)j.
Fréquent: ataxiek, convulsions, hémiparésie, neuropathie périphériquel, myoclonie.
Occasionnel: état de mal épileptique, quadriplégie, oedème de la moelle épinière, myélite, paralysie facialem, dyscalculie.
Affections cardiaques:
Très fréquent: tachycardie (21%)n, arythmie (19%)o.
Fréquent: insuffisance cardiaquep.
Occasionnel: arrêt cardiaque.
Affections vasculaires:
Très fréquent: hypotension (21%)g, hypertension (10%).
Fréquent: thromboser.
Occasionnel: syndrome d'hyperperméabilité capillaire.
Affections respiratoires, thoraciques et médiastinales:
Très fréquent: toux (26%).
Fréquent: hypoxiet, épanchement pleural, œdème pulmonaire, dyspnéeu, rhinorrhée (y compris rhinite allergique).
Occasionnel: défaillance pulmonairev.
Affections gastro-intestinales:
Très fréquent: vomissement (18%), diarrhée (34%)w, constipation (23%), douleur abdominale (18%)x, nausée (33%).
Fréquent: dysphagie, bouche sèchey.
Affections hépatobiliaires:
Très fréquent: transaminases augmentées (18%)z.
Fréquent: hyperbilirubinémieaa.
Affections de la peau et du tissu sous-cutané:
Très fréquent: rash (y compris rash, rash au site d'administration, dermatite, dermatite allergique, dermite bulleuse, érythème, prurit, rash érythémateux, rash maculeux, rash maculopapuleux, rash prurigineux, rash pustuleux, syndrome de Stevens-Johnson, urticaire) (15%).
Affections musculosquelettiques et du tissu conjonctif:
Très fréquent: dysfonction motrice (16%)bb, douleur musculosquelettique (38%)cc.
Occasionnel: rhabdomyolyse.
Affections du rein et des voies urinaires:
Fréquent: atteinte de la fonction rénaledd.
Troubles généraux et anomalies au site d'administration:
Très fréquent: fièvre (23%)ee, oedème (16%)ff, fatigue (44%)gg, frissons (12%).
Fréquent: douleurs.
Occasionnel: syndrome de défaillance multiviscérale.
Troubles oculaires
Fréquent: défauts visuelshh.
Notes:
* Fréquence basée sur des anomalies biologiques de grade 3 ou plus
# L'hyperuricémie a été constatée à l'aide d'une analyse combinée de 232 patients adultes dans ZUMA-1 et ZUMA-5
a. La coagulopathie inclut coagulopathie, fibrinogène sanguin diminué, fibrinogène sanguin augmenté, coagulation intravasculaire disséminée, hypofibrinogénémie, INR (international normalised ratio) augmenté, prothrombine diminuée, temps de prothrombine allongé
b. Immunoglobuline G sanguine diminuée, immunoglobulines diminuées incluent hypogammaglobulinémie
c. La diminution de l'appétit inclut appétit diminué, hypophagie
d. La déshydratation inclut déshydratation, hypovolémie,
e. Le délire inclut délire, agitation, idée délirante, désorientation, hallucination, instabilité psychomotrice
f. Les troubles affectifs incluent comportement impulsif, manie, humeur modifiée, attaques de panique
g. L'encéphalopathie inclut encéphalopathie, agraphie, état modifié de conscience, amnésie, aphasie, aphonie, apraxie, trouble cognitif, état confusionnel, diminution du niveau de conscience, perturbations de l'attention, dysarthrie, dysgraphie, dyskinésie, dyspraxie, hypersomnie, syndrome de neurotoxicité liée aux cellules effectrices de l'immunité, léthargie, leucoencéphalopathie, perte de conscience, atteinte de la mémoire, détérioration mentale, modifications de l'état mental, encéphalopathie métabolique, neurotoxicité, élocution lente, somnolence, trouble de la parole, stupeur, encéphalopathie toxique
h. Le tremblement inclut tremblement, titubation de la tête
i. Les céphalées incluent céphalées, gêne de la tête, céphalée de tension
j. Les sensations vertigineuses incluent sensations vertigineuses, sensation vertigineuse posturale, présyncope, syncope, vertige
k. L'ataxie inclut ataxie, trouble de l'équilibre, troubles de la démarche
l. La neuropathie périphérique inclut neuropathie périphérique, allodynie, radiculopathie cervicale, hyperesthésie, hypoesthésie, radiculopathie lombaire, paresthésie, parosmie, neuropathie périphérique motrice, neuropathie périphérique sensitive, paralysie du nerf sciatique poplité externe
m. La paralysie faciale inclut paralysie faciale
n. La tachycardie inclut tachycardie, syndrome de tachycardie en posture orthostatique, tachycardie sinusale
o. L'arythmie inclut arythmie, fibrillation auriculaire, flutter auriculaire, bloc auriculoventriculaire, bloc auriculoventriculaire de premier grade, bradycardie, bloc de branche droit, intervalle QT prolongé à l'électrocardiogramme, inversion de l'onde T dans l'électrocardiogramme, extrasystoles, fréquence cardiaque augmentée, fréquence cardiaque irrégulière, bradycardie sinusale, extrasystoles supraventriculaires, tachycardie supraventriculaire, arythmie ventriculaire, extrasystoles ventriculaires, tachycardie ventriculaire
p. L'insuffisance cardiaque inclut insuffisance cardiaque, insuffisance ventriculaire gauche aiguë, fraction d'éjection diminuée, cardiomyopathie provoquée par le stress
q. L'hypotension inclut hypotension, hypotension diastolique, hypoperfusion, hypotension orthostatique
r. La thrombose inclut thrombose, thrombose de la veine axillaire, thrombose de la veine brachiocéphalique, thrombose veineuse profonde, occlusion d'un dispositif médical, embolie, thrombose de la veine jugulaire, embolie artérielle périphérique, ischémie périphérique, embolie pulmonaire, thrombose de la veine splénique, thrombose de la veine sous-clavière, thrombose dans un dispositif médical, occlusion vasculaire
s. La toux inclut toux, toux productive, syndrome de toux d'origine des voies aériennes supérieures
t. L'hypoxie inclut hypoxie, saturation en oxygène diminuée
u. La dyspnée inclut dyspnée, dyspnée d'effort
v. L'insuffisance respiratoire inclut insuffisance respiratoire, insuffisance respiratoire aiguë
w. La diarrhée inclut diarrhée, colite, entérite
x. Les douleurs abdominales incluent douleurs abdominales, gêne abdominale, douleur abdominale basse, douleur abdominale haute, abdomen sensible (à la pression), dyspepsie, gêne épigastrique
y. La bouche sèche inclut bouche sèche, lèvres sèches
z. L'élévation des transaminases inclut élévation des transaminases, alanine aminotransférase augmentée, aspartate aminotransférase augmentée, enzymes hépatiques augmentées,
aa. L'hyperbilirubinémie augmentée inclut hyperbilirubinémie, bilirubine sanguine augmentée
bb. La dysfonction motrice inclut dysfonction motrice, contractions musculaires involontaires, rigidité musculaire, contractures musculaires, spasticité musculaire, claquage de muscle, tensions musculaires, contractions musculaires, faiblesse musculaire
cc. Les douleurs musculosquelettiques incluent douleurs musculosquelettiques, arthralgie, arthrite, dorsalgies, douleurs osseuses, douleurs du flanc, douleurs inguinales, douleurs musculosquelettiques du thorax, myalgie, cervicalgies, arthrose, extrémités douloureuses
dd. L'atteinte de la fonction rénale inclut atteinte aiguë de la fonction rénale, créatinine sanguine augmentée, insuffisance rénale
ee. La fièvre inclut hyperthermie, fièvre
ff. L'œdème inclut œdème, œdème de la face, œdème généralisé, œdème localisé, œdème génital, œdème périphérique, gonflement périphérique, gonflement
gg. La fatigue inclut fatigue, asthénie, diminution de l'activité, malaise
hh. Les défauts visuels incluent défauts visuels, hémianopsie, vision trouble, baisse de l'acuité visuelle
Tableau 3: Effets médicamenteux indésirables dans ZUMA-7 avec une différence de fréquence entre les bras de traitement de ≥10%
|
Classe de systèmes d'organes (SOC)
Effet indésirable
|
YESCARTA
(n = 170)
|
Traitement standard
(n = 168)
| |
Infections et infestations
| |
Infections virales
|
15%
|
5%
| |
Affections hématologiques et du système lymphatique
| |
Leucopénie de grade ≥3#
|
95%
|
56%
| |
Lymphopénie de grade ≥3#
|
99%
|
68%
| |
Neutropénie de grade ≥3#
|
94%
|
51%
| |
Thrombopénie de grade ≥3#
|
26%
|
63%
| |
Affections du système immunitaire
| |
Syndrome de relargage des cytokines
|
92%
|
0%
| |
Immunoglobulines diminuéesa
|
11%
|
1%
| |
Troubles du métabolisme et de la nutrition
| |
Hyponatrémie de grade ≥3
|
12%
|
2%
| |
Affections du système nerveux
| |
Encéphalopathieb
|
49%
|
8%
| |
Tremblementc
|
25%
|
1%
| |
Affections respiratoires, thoraciques et médiastinales
| |
Touxd
|
27%
|
11%
| |
Affections gastro-intestinales
| |
Nausée
|
34%
|
69%
| |
Vomissement
|
15%
|
33%
| |
Constipation
|
20%
|
35%
| |
Affections hépatobiliaires
| |
Transaminases augmentéese
|
21%
|
11%
| |
Affections du rein et des voies urinaires
| |
Atteinte de la fonction rénalef
|
9%
|
19%
| |
Troubles généraux et anomalies au site d'administration
| |
Fatigueg
|
44%
|
57%
| |
Fièvreh
|
16%
|
26%
|
Dans le tableau 3 ne figurent que les cytopénies qui (i) ont entraîné des atteintes cliniques secondaires nouvelles ou aggravées ou (ii) ont nécessité un traitement ou (iii) ont conduit à un ajustement du traitement en cours.
# Fréquence basée sur les paramètres biologiques
a. Les immunoglobulines diminuées incluent hypogammaglobulinémie, immunoglobulines G sanguines diminuées
b. L'encéphalopathie inclut encéphalopathie, état modifié de conscience, amnésie, aphasie, apraxie, trouble cognitif, état confusionnel, diminution du niveau de conscience, perturbations de l'attention, dysarthrie, dysgraphie, dyskinésie, dyspraxie, hypersomnie, léthargie, leucoencéphalopathie, perte de conscience, atteinte de la mémoire, détérioration mentale, modifications de l'état mental, encéphalopathie métabolique, élocution lente, somnolence, stupeur, encéphalopathie toxique.
c. Le tremblement inclut tremblement, titubation de la tête
d. La toux inclut toux, toux productive, syndrome de toux d'origine des voies aériennes supérieures.
e. Les transaminases augmentées incluent transaminases augmentées, enzyme hépatique augmentée, alanine aminotransférase augmentée, aspartate aminotransférase augmentée
f. L'atteinte de la fonction rénale inclut créatinine sanguine augmentée, atteinte aiguë de la fonction rénale
g. La fatigue inclut fatigue, asthénie, malaise
h. La fièvre inclut la pyrexie
Description de certains effets indésirables issus des essais ZUMA-1, ZUMA-7 et ZUMA-5
Syndrome de relargage cytokinique:
Un CRS est survenu chez 92% des patients dans ZUMA-1 et ZUMA-7. Un CRS de grade 3 ou supérieur (sévère, mettant en jeu le pronostic vital, ou fatal) est survenu chez huit pour cent (8%) des patients. Le délai médian de survenue était de 3 jours (intervalle: de 1 à 12 jours) et la durée médiane était de 7 jours (avec un intervalle de 2 à 29 jours, à l'exception d'une observation aberrante de 58 jours). 99% des patients ont eu une résolution de leur CRS.
Un CRS est survenu chez 78% des patients dans ZUMA-5. Un CRS de grade 3 ou supérieur (sévère, mettant en jeu le pronostic vital, ou fatal) est survenu chez six pour cent (6%) des patients. Le délai médian de survenue était de 4 jours (intervalle: de 1 à 15 jours) et la durée médiane était de 6 jours (avec un intervalle de 1 à 27 jours). Nonante-neuf pour cent (99%) des patients ont eu une résolution de leur CRS. Un cas fatal de CRS est survenu dans ZUMA-5.
Les effets indésirables les plus fréquents (≥20%) pouvant être associés au CRS étaient la fièvre (89%), l'hypotension (50%), la fatigue (50%), la tachycardie (47%), les céphalées (43%), la diarrhée (37%), les nausées (37%), les frissons (30%), l'hypoxie (25%) et les vomissements (23%). Les effets indésirables sévères pouvant être associés au CRS sont les suivants: fièvre (12%), hypotension (5%), hypoxie (2%), arythmie (3%), insuffisance cardiaque (2%), fatigue (2%), céphalées (2%), tachycardie (2%), arrêt cardiaque (1%) et dyspnée (1%). Pour les recommandations en matière de surveillance et de traitement voir «Mises en garde et précautions».
Effets indésirables neurologiques:
Des effets indésirables neurologiques sont survenus chez 62% des patients dans ZUMA-1 et ZUMA-7. Des effets indésirables de Grade 3 ou supérieur (sévères ou mettant en jeu le pronostic vital) sont survenus chez vingt-cinq (25%) des patients. Chez 75% des patients les effets indésirables neurologiques sont survenus dans les 7 premiers jours après la perfusion. Le délai médian de survenue était de 6 jours (intervalle 1 à 133 jours). La durée médiane était de 10 jours avec un intervalle de 1 à 817 jours, les symptômes s'étant atténués chez 66% des patients dans les 3 semaines après la perfusion. Les effets indésirables neurologiques se sont résolus chez la majorité des patients, à l'exception de 10 patients (3,6% des patients traités par YESCARTA dans ZUMA-1 et ZUMA-7). Les effets indésirables neurologiques n'avaient pas encore disparu au moment du décès, mais les patients sont décédés pour des raisons qui n'étaient pas en rapport avec ces derniers.
Des effets indésirables neurologiques sont survenus chez 56% des patients dans ZUMA-5. Des effets indésirables de Grade 3 ou supérieur (sévères ou mettant en jeu le pronostic vital) sont survenus chez quinze (15%) des patients. Chez 66% des patients les effets indésirables neurologiques sont survenus dans les 7 premiers jours après la perfusion. Le délai médian de survenue était de 7 jours (intervalle: 1 à 177 jours). La durée médiane était de 14 jours, les symptômes s'étant atténués chez 60% des patients dans les 3 semaines après la perfusion.
Les effets indésirables les plus fréquents (≥5%) associés aux effets indésirables neurologiques incluaient encéphalopathie (51%), céphalées (33%), tremblements (28%), sensation vertigineuse (22%), délire (13%), insomnie (13%), neuropathie périphérique (9%), anxiété (7%), et ataxie (6%). Les effets indésirables sévères (≥1%), dont encéphalopathie (18%), délire (2%) et tremblements (1%), ont été rapportés chez les patients traités par YESCARTA.
D'autres effets indésirables neurologiques ont été rapportés moins fréquemment dans les essais cliniques parmi lesquels la dysphagie (3%), la myélite (0,2%) et la quadriplégie (0,1%).
Un patient dans ZUMA-1 a présenté un épisode d'hémorragie intracrânienne fatal, qui a coïncidé avec un CRS, une neurotoxicité, une thrombopénie grave, une administration d'héparine pour la prophylaxie de la thrombose veineuse profonde (TVP) et une septicémie bactérienne persistante. La cause du décès était un sepsis attribué à la chimiothérapie de conditionnement.
Les effets indésirables rapportés après mise sur le marché comprennent l'état de mal épileptique (0,3%), l'œdème de la moelle épinière, réactions liées à la perfusion, , l'hémorragie (hémorragie gastro-intestinale, hémorragie cérébrale, hémorragie pulmonaire, hémorragie intracrânienne, choc hémorragique, hémorragie, cystite hémorragique, hémorragie sous-arachnoïdienne), les tumeurs malignes à cellules T et l'ICANS, qui ont été associés à une toxicité neurologique.
Pour les recommandations en matière de surveillance et de traitement voir «Mises en garde et précautions».
Infections et neutropénie fébrile:
Des neutropénies fébriles ont été observées chez 10% des patients après la perfusion de YESCARTA. Des infections sont survenues chez 48% des patients. Des infections de Grade 3 ou supérieur (sévères, mettant en jeu le pronostic vital, ou fatales) sont survenues chez 19% des patients. Des infections à des agents pathogènes non spécifiés, des infections bactériennes et des infections virales de Grade 3 ou supérieur sont survenues chez 12%, 6% et 4% des patients. Le site le plus fréquent d'infections à agents pathogènes non spécifiés était les voies respiratoires. Pour les recommandations en matière de surveillance et de traitement voir «Mises en garde et précautions».
Cytopénies prolongées:
Des neutropénies, des anémies et des thrombopénies de Grade 3 ou supérieur sont survenues chez 68%, 32% et 24% des patients respectivement. Des neutropénies (y compris neutropénie, numération des neutrophiles plasmatiques diminuée et neutropénie fébrile), des thrombopénies et des anémies, de Grade 3 ou supérieur, qui duraient jusqu'au Jour 30 ou plus longtemps, sont survenues chez 27%, 12% et 6% des patients. Pour les recommandations en matière de traitement voir «Mises en garde et précautions».
Hypogammaglobulinémie:
Une hypogammaglobulinémie a été rapportée chez 15% des patients traités par YESCARTA. En cumul, 36 (33%) des 108 patients dans ZUMA-1 ont reçu un traitement intraveineux par immunoglobulines jusqu'au moment de l'analyse à 24 mois, et 28 (16%) des 170 patients traités par YESCARTA dans ZUMA-7 ont reçu un traitement intraveineux par immunoglobulines jusqu'au moment de l'analyse principale. Dans ZUMA-5, 35 (28%) des 124 patients ont reçu un traitement intraveineux par immunoglobulines jusqu'au moment de l'analyse de suivi à 24 mois. Pour les recommandations en matière de traitement voir «Mises en garde et précautions».
Immunogénicité
L'immunogénicité de YESCARTA a été évaluée au moyen d'un test d'immunadsorption couplé à des enzymes (Enzyme-Linked Immunosorbent Assay, ELISA) pour détecter les anticorps fixant le FMC63, l'anticorps d'origine du CAR Anti-CD19, suivi d'un test de confirmation basé sur cellules. Les patients ont été suivis à l'inclusion, avant la perfusion et jusqu'à 1 an après la perfusion. Aucun des patients traités par YESCARTA n'avait développé d'anticorps de novo après la perfusion.
Onze sur 278 patients (4%) ont eu un résultat positif pour les anticorps anti-FMC63 avant d'être traités par YESCARTA dans ZUMA-1 et ZUMA-7, et un patient (1%) de l'étude ZUMA-7 qui avait eu un résultat négatif au test avant le traitement, a eu un résultat positif au test après le traitement, par dépistage ELISA. Les résultats d'un test cellulaire de confirmation, s'appuyant sur la partie extracellulaire correctement repliée et exprimée de CAR (scFv, région charnière et espaceur) ont démontré que tous les patients traités par YESCARTA ayant eu un résultat positif au dépistage ELISA étaient négatifs aux anticorps à tous les points d'évaluation testés. Il n'existe aucune donnée probante indiquant que la cinétique de l'expansion initiale et de la persistance de cellules CAR-T ou la sécurité ou l'efficacité de YESCARTA, aient été altérées chez ces patients.
Dans ZUMA-5, 14 des 124 patients atteints de FL ont eu un résultat préliminaire positif pour les anticorps dans le test ELISA avant d'être traités par YESCARTA, et 3 sujets ayant eu des résultats négatifs au test ELISA avant le traitement ont eu des résultats positifs après le traitement. Les résultats d'un test cellulaire de confirmation ont démontré que tous les patients dans ZUMA-5 traités par YESCARTA et ayant eu un résultat positif au test ELISA étaient négatifs aux anticorps avant et après le traitement. Aucun impact de ces résultats n'était détecté par rapport aux anticorps sur la sécurité et l'efficacité clinique.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageIl n'existe aucune donnée concernant les signes de surdosage avec YESCARTA.
Propriétés/EffetsCode ATC
L01XL03
Mécanisme d'action
YESCARTA, un médicament d'immunothérapie constitué de cellules T d'immunothérapie autologues génétiquement modifiées, se lie aux cellules cancéreuses et aux cellules B normales exprimant le CD19. Suite à la liaison des cellules CAR T anti-CD19 avec les cellules cibles exprimant le CD19, les domaines de co-stimulation CD28 et CD3-zéta activent la cascade de signalisation qui conduit dans les cellules T, à l'activation, la prolifération, à l'acquisition de fonctions effectrices et à la sécrétion des cytokines et des chimiokines inflammatoires. Cette cascade d'événements conduit à l'apoptose et à la nécrose des cellules cibles exprimant le CD19.
Pharmacodynamique
Après la perfusion de YESCARTA, la réponse pharmacodynamique a été évaluée en mesurant l'élévation transitoire des cytokines, des chimiokines et d'autres molécules dans le sang à 4 semaines d'intervalle. Les taux de cytokines et de chimiokines telles que les interleukines (IL) IL-6, IL-8, IL-10, IL-15, le TNF-α, l'IFN-γ et sIL2Rα ont été analysés. Un pic d'élévation a été observé au cours des 14 premiers jours après la perfusion et les taux étaient généralement revenus aux valeurs initiales en 28 jours.
En raison de l'effet on-target, off-tumour de YESCARTA, une aplasie de cellules B est attendue sur un certain intervalle après le traitement. Parmi les 73 patients atteints de LDGCB et de LMPGB dans ZUMA-1 pour lesquels des échantillons évaluables étaient disponibles au début de l'étude, 40% présentaient des cellules B détectables; l'aplasie de cellules B observée au début chez la majorité des patients a été attribuée à des traitements antérieurs. Après le traitement par YESCARTA, la proportion de patients présentant des cellules B détectables a diminué: 20% d'entre eux présentaient des cellules B détectables à 3 mois et 22% d'entre eux présentaient des cellules B détectables à 6 mois.
L'initiation d'une régénération des cellules B a été constatée pour la première fois à 9 mois, alors que 56% des patients présentaient des cellules B détectables. Cette tendance de régénération des cellules B s'est poursuivie sur la durée puisque 64% des patients présentaient des cellules B détectables à 18 mois et 77% des patients présentaient des cellules B détectables à 24 mois.
Parmi les 141 patients dans ZUMA-7 avec des échantillons évaluables au début de l'étude, 57% présentaient des cellules B détectables. Après le traitement par YESCARTA, la proportion des patients présentant des cellules B détectables a diminué: 38% présentaient des cellules B détectables au mois 3 et 41% présentaient des cellules B détectables à 6 mois. L'initiation de la régénération des cellules B a été constatée pour la première fois à 9 mois, alors que 58% des patients présentaient des cellules B détectables. Cette tendance de régénération des cellules B s'est poursuivie sur la durée puisque 64% des patients présentaient des cellules B détectables à 18 mois et 85% des patients présentaient des cellules B détectables à 24 mois.
Parmi les 113 patients dans ZUMA-5 atteints de LF ayant des échantillons évaluables à l'inclusion, 75% des patients présentaient des lymphocytes B détectables. Après le traitement par YESCARTA, la proportion des patients présentant des lymphocytes B détectables a diminué: 40% des patients présentaient des lymphocytes B détectables à 3 mois. La restauration des lymphocytes B a été observée au cours du temps, 61% des patients présentant des lymphocytes B détectables à 24 mois.
Il est important de noter que les patients n'ont pas eu besoin d'être suivis après la progression de la maladie; par conséquent, la majorité des patients dont les échantillons étaient évaluables étaient des répondeurs.
Efficacité clinique
Étude clinique ZUMA, phases 1 et 2 (LDGCB récidivant ou réfractaire, LMPCB et LDGCB résultant d'un lymphome folliculaire, après au moins deux lignes de traitement systémique)
Un total de 108 patients (7 patients inclus dans la phase 1 et 101 patients inclus dans la phase 2) atteints de lymphome non Hodgkinien (LNH) à cellules B agressif r/r ont été traités par YESCARTA dans une étude de phase 1/2 en ouvert, multicentrique, monobras.
D'après la classification OMS de 2008, qui était valide au moment de l'étude, l'efficacité a été évaluée sur 101 patients inclus dans la phase 2, y compris des patients atteints de LDGCB (N = 77), de LMPGB (N = 8) et de LDGCB qui résultaient histologiquement d'un lymphome folliculaire (N = 16). D'après la classification actuelle de l'OMS 2016:
Le LDGCB dans ZUMA-1 incluait des patients atteints de LDGCB non spécifiés, d'autres sous-types de LDGCB et de LBHG. Ceci est basé sur l'analyse rétrospective, post-hoc d'une partie des patients par «independent pathology review». Quarante-sept patients étaient évaluables pour le statut MYC, BCL-2, et BCL-6. Trente patients présentaient un LDGCB double expresseur (surexpression des protéines MYC et BCL-2); 5 patients étaient atteints d'un LBHG avec réarrangement des gènes MYC, BCL-2 ou BCL-6 (mutation de 2 gènes (double hit) ou de 3 gènes (triple hit)); 2 patients étaient atteints d'un LBHG non spécifié. En raison du nombre restreint de patients atteints de LBHG, aucune conclusion ne peut être établie par rapport à l'efficacité clinique au sein de cette population de patients. Soixante-six patients étaient évaluables pour la détermination des cellules B d'origine (cellules B germinales [GCB] ou cellules B activées [ABC]). Parmi ces patients, 49 présentaient un type GCB et 17 patients présentaient un type ABC.
Les patients éligibles étaient âgés d'au moins 18 ans et présentaient une maladie réfractaire, définie comme maladie progressive (progressive disease, PD) ou comme maladie stable (stable disease, SD) comme meilleure réponse à la dernière ligne de traitement utilisée ou présentant une progression de la maladie au cours des 12 derniers mois après une greffe de cellules souches [GCS] autologues . Les patients qui sont réfractaires à une chimiothérapie ou en récidive après au moins deux lignes de traitement systémique sont généralement inéligibles à une greffe de cellules souches hématopoïétiques (GSCH).
Les patients devaient avoir reçu au moins un traitement antérieur par des anticorps anti-CD20 ainsi qu'un régime à base d'anthracycline. Les patients présentant un lymphome du SNC, ou ayant fait l'objet d'une greffe de cellules souches hématopoïétiques allogéniques ou d'un traitement antérieur par CAR T anti-CD19 ou un autre traitement à base de cellules T génétiquement modifiées ont été exclus. Les patients ayant des antécédents de troubles du SNC (p.ex. convulsions ou ischémie vasculaire cérébrale), une fraction d'éjection cardiaque inférieure à 50%, ou une saturation en oxygène en air ambiant inférieure à 92% ou une maladie autoimmune nécessitant un traitement immunosuppresseur systémique étaient exclus. La durée médiane du suivi était de 27,1 mois (toujours en cours). La population en ITT était définie comme l'ensemble des patients ayant eu une leucaphérèse, et la population en mITT comme l'ensemble des patients ayant reçu YESCARTA.
Données démographiques de la population intent-to-treat (mITT) modifiée
L'âge médian de la population de l'étude mITT était 58 ans (intervalle: 23 à 76 ans); 67% étaient des hommes, 86% étaient caucasiens, 3% étaient d'origine asiatique et 4% étaient afro-américains. Le statut ECOG au début de l'étude correspondait pour 42% à ECOG 0 et pour 58% à ECOG 1. Le nombre médian de traitements précédents était 3 (intervalle: 1 à 10); pour 76% des patients, la maladie était réfractaire à 2 lignes de traitement précédentes ou plus, et 21% des patients étaient en récidive dans l'année suivant la greffe autologue de cellules souches [GCS] autologues. 46% des patients avaient un indice pronostique international de 3/4, et 85% des patients étaient atteints d'une maladie de stade III/ IV.
Données démographiques de la population intent-to-treat (ITT)
L'âge médian de la population de l'étude ITT était 58 ans (intervalle: 23 à 76 ans); 69% étaient des hommes, 85% étaient caucasiens, 4% étaient d'origine asiatique et 4% étaient afro-américains. Le statut ECOG au début de l'étude correspondait pour 41% à ECOG 0 et pour 59% à ECOG 1. Le nombre médian de traitements précédents était 3 (intervalle: 1 à 10); pour 77% des patients, la maladie était réfractaire à 2 lignes de traitement précédentes ou plus, et 20% des patients étaient en récidive dans l'année suivant la GCSA. 46% des patients avaient un indice pronostique international de 3/4, et 85% des patients étaient atteints d'une maladie de stade III/ IV.
YESCARTA a été administré en une perfusion unique à une dose cible de 2 x 106 cellules CAR T anti-CD19/kg à l'issue d'un schéma de chimiothérapie lymphodéplétive par 500 mg/m2 de cyclophosphamide par voie intraveineuse et 30 mg/m2 de fludarabine par voie intraveineuse; la lymphodéplétion a eu lieu les 5ème, 4ème et 3ème jours avant le traitement par YESCARTA. L'ensemble des 108 patients traités par YESCARTA dans l'étude ZUMA-1 (phases 1 et 2) ont reçu une chimiothérapie lymphodéplétive. Tous les patients avaient un nombre initial de globules blancs ≥1 x 103/μl (à savoir avant la chimiothérapie lymphodéplétive). Seuls les patients présentant un nombre absolu de neutrophiles ≥1000/μl, un nombre absolu de lymphocytes ≥100/μl et un taux de plaquettes ≥75 000/μl au départ ont été inclus dans l'étude. Une chimiothérapie en relais entre la leucaphérèse et la chimiothérapie lymphodéplétive n'était pas autorisée. Tous les patients ont été hospitalisés dans le but de surveiller et prendre en charge des effets indésirables survenant pendant au moins 7 jours après la perfusion de YESCARTA.
Phase 2 de ZUMA-1
Parmi les 111 patients qui ont eu une leucaphérèse, 101 ont reçu YESCARTA. Neuf patients n'ont pas été traités, en raison, principalement, de progression de la maladie ou d'événements indésirables sévères après le recrutement et avant l'administration des cellules. Sur les 111 patients, un patient n'a pas reçu le médicament en raison d'un problème de fabrication. Le délai médian entre la leucaphérèse et la réception du médicament était de 17 jours (intervalle: 14 à 51 jours), et le délai médian entre la leucaphérèse et la perfusion était de 24 jours (intervalle: 16 à 73 jours). La dose médiane était de 2,0 x 106 cellules CAR T anti-CD19/kg.
Le critère de jugement principal était le taux de réponse objective (Objective Response Rate, ORR) évalué par les médecins-investigateurs. Les critères d'évaluation secondaires incluaient le taux de réponse objective (ORR) évalué par un comité d'évaluation indépendant, la durée de la réponse (Duration of Response, DOR), la survie sans progression (PFS), la survie globale (Overall Survival, OS) et la sévérité des effets indésirables. Il était attendu que le ORR devait être testé chez les 92 premiers patients traités et être significativement plus élevé que le taux préspécifié de 20% (p < 0,0001).
Dans l'analyse préliminaire, basée sur la population en intention de traiter modifiée (mITT) (suivi minimum de 6 mois) le ORR a été de 82%, et le taux de réponse complète (Complete Response, CR) était de 54%, selon les médecins-investigateurs (critère de jugement principal). Dans l'analyse actualisée (suivi minimum de 12 mois), le ORR était de 83%, et le taux de CR était de 58% selon les médecins-investigateurs. Dans l'analyse de suivi à 24 mois, le ORR était de 83% et le taux de RC était de 58% selon les médecins-investigateurs. Les résultats d'efficacité sont résumés ci-dessous, dans le tableau 4. La valeur médiane OS au sein de la population en ITT était de 17,4 mois (IC à 95% de 11,6 NE). Les taux de OS à 12 et 24 mois étaient respectivement de 59,5% et 47,7%. La valeur médiane de l'OS n'a pas encore été atteinte au sein de la population en mITT parmi les 50 événements/101 patients observés. Les taux de OS à 12 et 24 mois étaient respectivement de 60,4% et 50,5%. Dans une analyse à 36 mois, la valeur médiane OS au sein de la population en mITT (101 patients) était de 25,8 mois. Dans une analyse à 60 mois, les estimations de Kaplan-Meier des taux de OS à 3 ans, 4 ans et 5 ans étaient de 47%, 44% et 43% respectivement.
Tableau 4: Résumé des résultats d'efficacité pour la phase 2 de ZUMA-1 (analyse à 24 mois)
|
|
Tous les patients ayant eu une leucaphérèse (ITT)
Cohorte 1 + 2
(N = 111)
|
Tous les patients traités
mITT
Cohorte 1 + 2
(N = 101)
| |
|
Évaluation d'une commission d'examen indépendante
|
Évaluation de médecins-investigateurs
|
Évaluation d'une commission d'examen indépendante
|
Évaluation de médecins-investigateurs
| |
ORR (%) [IC à 95%]
|
68 (58, 76)
|
77 (69, 85)
|
74 (65, 82)
|
83 (74, 90)
| |
CR (%)
|
50
|
55
|
54
|
58
| |
PFS (mois) [IC à 95%]
|
9,5 (6,1, 15,4)
|
6,2 (4,0, 12,4)
|
9,1 (5,7, n.e.)
|
5,9 (3,3, 15,0)
| |
DOR,a, médiane (IC à 95%) en mois
|
n.e. (10,9, n.e.)
|
9,0 (3,9, n.e.)
|
n.e. (10,9, n.e.)
|
9,0 (3,9, n.e.)
| |
DOR, CR, médiane (IC à 95%) en mois
|
n.e. (n.e., n.e.)
|
non disponible
|
n.e. (n.e., n.e.)
|
n.e. (12,9, n.e.)
| |
DOR, PR, médiane (IC à 95%) en mois
|
2,1 (1,3, 11,1)
|
non disponible
|
2,1 (1,3, 11,1)
|
1,9 (1,3, 2,1)
| |
Suivi médian (mois)
|
27,1
| |
Suivi minimal (mois)
|
22,9
| |
OS, médiane (mois) [IC à 95%]
|
17,4 (11,6, n.e.)
|
n.e. (12,8, n.e.)
| |
OS à 6 mois (%) [IC à 95%]
|
81,1 (72,5, 87,2)
|
79,2 (69,9; 85,9)
| |
OS à 12 mois (%) [IC à 95%]
|
59,5 (49,7, 67,9)
|
60,4 (50,2, 69,2)
| |
OS à 24 mois (%) [IC à 95%]
|
47,7 (38,2, 56,7)
|
50,5 (40,4, 59,7)
|
IC, intervalle de confiance; CR, réponse complète; DOR, durée de la réponse; ITT, intention de traiter; mITT, intention de traiter modifiée; n.e. = non estimable; ORR, taux de réponse objective; OS, survie globale; PR, réponse partielle.
a La durée de la réponse et la PFS ont été censurées au moment de la GCSH pour les patients ayant reçu une GCSH alors qu'ils continuaient à répondre
Note: la mITT était définie par tous les patients qui recevaient au moins une dose de YESCARTA de 1x106 cellules CAR-T/kg
Étude clinique ZUMA-7, phase 3, (LDGCB r/r et LBHG r/r)
L'efficacité et la sécurité de YESCARTA chez les patients adultes atteints de LDGCB r/r et LBHG r/r ont été démontrées dans une étude de phase 3 randomisée, en ouvert et multicentrique (ZUMA-7). La plupart des patients inclus avaient reçu le diagnostic d'un LDGCB non autrement spécifié/non classifiable davantage (69%) et d'un lymphome B de haut grade (LBHG) (0% à LBHG non autrement spécifié et 16% à réarrangement des gènes MYC / BCL-2 / BCL-6 (y compris double hit ou triple hit)) selon l'évaluation du laboratoire central. 46 patients (13%) ont été catégorisés en tant que «non confirmés» ou «manquants», et 10 patients (3%) ont été catégorisés en tant que «lymphome autre que LDGCB ou LBHG». Les patients ont été encore catégorisés selon les sous-groupes moléculaires (type cellule B de centre germinatif (58%) et type cellule B activée (7%) ainsi que «non classés» (9%)) et le statut d'expression de CD19 «oui» (77%) et «non» (7%) selon l'évaluation du laboratoire central. Tous les patients avaient reçu au préalable un traitement de première ligne avec une thérapie à base d'anthracycline et des anticorps monoclonaux anti-CD20, à moins que la tumeur ait été CD-20-négative. Au total, 359 patients ont été randomisés selon un rapport 1:1 pour recevoir une perfusion unique de YESCARTA ou recevoir le traitement standard (défini par 2 à 3 cycles de chimio-immunothérapie standard [R-ICE, R-DHAP ou R-DHAX, R-ESHAP ou R-GDP] suivie d'un traitement à haute dose [THD] et de greffe de cellules souches [GCS] autologue chez des patients dont la maladie répondait au traitement). La randomisation a été stratifiée 1) selon la réponse au traitement de première ligne (réfractaire primaire vs récidive ≤6 mois vs récidive > 6 et ≤12 mois) et selon l'aaIPP («age-adjusted international prognostic index») 0 / 1 vs 2 / 3. Les raisons de l'exclusion de l'étude étaient le traitement antécédent par greffe autologue de cellules souches hématopoïétiques (CSH), le lymphome primaire au niveau du SNC ou les métastases cérébrales, les cellules malignes détectables dans le liquide céphalorachidien et un indice de performance ECOG (Eastern Cooperative Oncology Group) de 2 ou plus. Les patients présentant des infections actives ou graves ont été exclus, toutefois les patients présentant des infections des voies urinaires simples et une pharyngite bactérienne non compliquée ont été autorisés s'ils répondaient à un traitement actif. Les patients atteints d'infections connues par le virus VIH, de l'hépatite B (HBsAG-positifs) ou de l'hépatite C (anti-VHC-positifs) ont été exclus. L'inclusion de patients après un traitement avec succès de l'hépatite B ou de l'hépatite C dans l'anamnèse était autorisée, si aucune charge virale n'était détectable dans la PCR quantitative et/ou le test d'acide nucléique.
Après une chimiothérapie lymphodéplétive, YESCARTA a été administré en une perfusion intraveineuse unique à une dose cible de 2 x 106 cellules CAR-T anti-CD19/kg (dose maximale: 2 x 108 cellules). Le schéma thérapeutique pour lymphodéplétion consistait en 500 mg/m² de cyclophosphamide par voie intraveineuse et 30 mg/m² de fludarabine par voie intraveineuse, tous deux administrés les 5e, 4e et 3e jours avant YESCARTA. Un traitement de transition d'attente non modificateur de la maladie, qui était limité aux corticostéroïdes, pouvait être administré entre la leucaphérèse et la chimiothérapie lymphodéplétive chez les patients ayant une charge tumorale élevée lors de la sélection.
Sur les 180 patients qui avaient été randomisés pour le traitement par YESCARTA, 178 ont été soumis à la leucaphérèse et 170 ont été traités par YESCARTA. Huit patients (4%) n'ont pas été traités après la leucaphérèse, notamment en raison de progression de la maladie, d'évènements indésirables graves ou de décès. Étant donné qu'une nouvelle administration pour un patient de l'étude clinique a nécessité un deuxième processus de fabrication, 171 charges ont été fabriquées au total. Le délai médian entre la leucaphérèse et la libération du produit était de 13 jours (intervalle: 10 à 24 jours) et le délai médian entre la leucaphérèse et la perfusion de YESCARTA était de 26 jours (intervalle: 16 à 52 jours). La dose médiane était de 2,0 x 106 cellules CAR-T anti-CD19/kg pour les patients ayant un poids corporel de < 100 kg (min: 1,0 x 106 cellules CAR-T anti-CD19/kg; max: 2,1 x 106 cellules CAR-T anti-CD19/kg). Pour les patients ayant un poids corporel de > 100 kg, la dose médiane était de 200 x 106 cellules CAR-T anti-CD19. 60 (33%) des patients traités ont reçu un traitement de transition d'attente par corticostéroïdes. Les 170 patients qui ont reçu YESCARTA ont tous été surveillés dans un établissement de santé pendant au moins 7 jours. Sur les 179 patients qui avaient été randomisés dans le bras de traitement standard de l'étude, 64 patients (36%) ont reçu une THD-GCSA, y compris 2 patients qui ont reçu une GCSA en dehors des spécifications du protocole.
Dans le groupe traitement standard 49 patients (41%) atteints de LDGCB et 9 patients (35%) atteints de LBHG ont reçu une GCSA-HD.
Dans la population globale de l'étude, l'âge médian était de 59 ans (intervalle: 21 à 81 ans); 66% étaient de sexe masculin et 83% étaient blancs. Septante-quatre pour cent des patients étaient atteints de LGCB réfractaire primaire, et 26% des patients étaient en récidive dans les 12 mois du traitement de première ligne. Les patients avaient un score IPI ajusté selon l'âge de 0-1 (55%) ou de 2-3 (45%) et un indice de performance ECOG de 0 (54%) ou de 1 (46%).
La durée réelle médiane de suivi était de 20,07 mois (intervalle: 0,59 à 37,75 mois) dans le groupe YESCARTA et de 18,23 mois (intervalle: 0,03 à 37,26 mois) dans le groupe de traitement standard.
Le critère d'évaluation principal était la survie sans événement (event-free survival, EFS), déterminée par une évaluation centralisée en aveugle. L'EFS était définie comme le temps entre la randomisation et la progression de la maladie selon la classification de Lugano (Cheson et al., 2014), le début d'un nouveau traitement du lymphome ou le décès de cause quelconque, en fonction de ce qui se produisait en premier. L'étude a montré une amélioration statistiquement significative de l'EFS chez les patients randomisés à YESCARTA en comparaison du traitement standard (HR: 0,398 [IC à 95%: 0,308; 0,514]). La survie à 24 mois sans évènement était de 40,5% [IC à 95%: 33,2; 47,7] dans le bras YESCARTA et de 16,3% [IC à 95%: 11,1; 22,2] dans le bras de traitement standard.
Les critères secondaires ORR et CR étaient significativement plus élevés chez les patients traités par YESCARTA, avec une différence d'OR de 33,1% [IC à 95%: 23,2; 42,1) et une différence de CR de 33% dans le bras YESCARTA (Tableau 5). La durée médiane de réponse était de 26,9 mois (intervalle: 0 à 29 mois) dans le bras YESCARTA, comparée à 8,9 mois (intervalle: 0 à 32 mois) dans le bras de traitement standard (HR: 0,736 [IC à 95%: 0,488; 1,108]). Au moment de l'analyse principale de l'EFS, la durée médiane de l'étude était de 24,9 mois.
L'analyse primairede la survie globale (OS), qui était un critère d'évaluation secondaire important, a été réalisée à la date définie dans le protocole d'étude, à savoir cinq ans à compter de l'inclusion du premier patient. Une amélioration statistiquement significative de l'OS avec YESCARTA par rapport au traitement standard a été démontréepar la réduction du risque de décès de 27,4% (HR: 0,726 [IC à 95%: 0,540; 0,977]). Avec des taux de OS à 48 mois estimés de 54,6% et 46,0% respectivement, l'OS médiane dans le bras YESCARTA n'a pas été atteinte comparée à 31,1 mois dans le bras de traitement standard. La durée médiane de l'étude est de 47,2 mois. Cinquante-sept pour cent (57%) des patients ont reçu une immunothérapie cellulaire après une absence de réponse au traitement standard ou après avoir rechuté après la randomisation au traitement standard.
Le résumé des résultats de l'efficacité dans la population globale est indiqué dans le tableau 5. La courbe de Kaplan-Meier de l'OS est illustrée dans la Figure 1.
Tableau 5: Résumé des résultats de l'efficacité pour ZUMA-7
|
|
YESCARTA
N = 180
|
Traitement standard
N = 179
| |
EFSa
| |
Nombre des évènements (%)
|
108 (60)
|
144 (80)
| |
Médiane, mois [IC à 95%]b
|
8,3 [4,5; 15,8]
|
2,0 [1,6; 2,8]
| |
Hazard Ratio stratifié [IC à 95%]
|
0,398 [0,308; 0,514]
| |
Valeur p du log-rank stratifiéec
|
< 0,0001
| |
OSf
| |
Nombre des évènements (%)
|
82 (46)
|
94 (53)
| |
OS médiane, mois [IC à 95%]b
|
Non atteinte (28,6; n. e.)
|
31,1 (17,1; n. e.)
| |
Hazard Ratio stratifié [IC à 95%]
|
0,726 (0,540; 0,977)
| |
Valeur p du log-rank stratifiéec,d
|
0,0168
| |
ORR (%) [IC à 95%]a
|
83 [77,1, 88,5]
|
50 [42,7, 57,8]
| |
Odds Ratio [IC à 95%]
|
5,31 [3,08, 8,90]
| |
Valeur p du test CMH stratifiéec
|
< 0,0001
| |
Taux de la réponse complète (%)
|
65 [57,6, 71,9]
|
32 [25,6, 39,8]
| |
Taux de la réponse partielle (%)
|
18 [13,0, 24,8]
|
18 [12,6, 24,3]
| |
PFSe
| |
Nombre des évènements (%)
|
96 (53)
|
103 (58)
| |
Médiane, mois [IC à 95%]b
|
14,7 [5,4, n. e.]
|
3,7 [2,9, 5,3]
| |
Hazard Ratio stratifié [IC à 95%]
|
|
0,490 [0,368, 0,652]
|
IC, intervalle de confiance; CMH, Cochran-Mantel-Haenszel; EFS, survie sans événement; n.e., non estimable; OS, survie globale; PFS, survie sans progression.
a Définie dans le cadre d'une évaluation centrale au moment de l'analyse principale de l'EFS.
b Méthode de Kaplan-Meier.
c Valeurs p unilatérales. Test du log-rank stratifié ou test CMH stratifié selon la réponse au traitement de première ligne (réfractaire primaire vs récidivant ≤6 mois après le traitement de première ligne vs récidivant > 6 et ≤12 mois après le traitement de première ligne) et ajusté à l'indice pronostique international en fonction de l'âge sur la base du traitement de deuxième ligne (0 à 1 vs 2 à 3).
d La valeur p est comparée à 0,0249, la limite d'efficacité unilatérale (seuil de signification) pour l'analyse principale de l'OS.
e Selon estimation de l'investigateur.
f Après évaluation au moment de l'analyse principale de l'OS (cinq ans à compter de l'inclusion du premier participant).
Une efficacité constante a été observée dans les sous-groupes, y compris réponse au traitement de première ligne, score IPI ajusté selon l'âge dans le traitement de deuxième ligne, indice de performance ECOG, âge, statut à double expression du lymphome, sous-type de maladie LBHG, statut CD19, symptômes B, implication de la rate, charge tumorale élevée et implication de la moelle osseuse.
Figure 1a. Courbe de Kaplan-Meier de la survie globale dans l'étude ZUMA-7 (ensemble complet d'analyses; analyse principale de la survie globale)
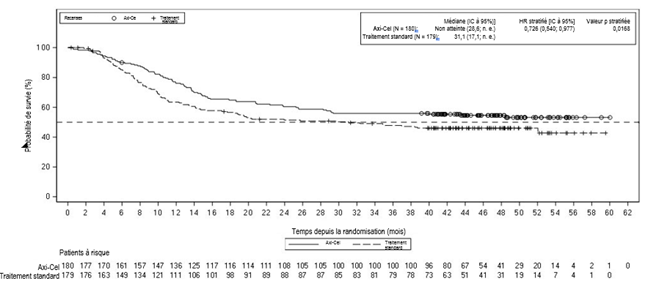
IC, intervalle de confiance; HR, Hazard Ratio; SOCT, traitement standard.
aLes patients qui ne répondaient pas au traitement standard ont pu recevoir, en dehors des spécifications du protocole, un traitement subséquent du lymphome, y compris un traitement par cellules CAR-T anti-CD19.
Traitement répété par YESCARTA
Au moment de l'analyse principale de l'OS, 10 patients (6%) au total ont reçu un traitement répété par YESCARTA. En raison du nombre restreint de patients, il n'est pas possible d'en tirer de conclusion en ce qui concerne l'efficacité et la sécurité.
Traitement subséquent du lymphome
Cinquante-sept pour cent (57%) des patients dans le bras de traitement standard ont reçu ensuite une immunothérapie cellulaire hors protocole (y compris 55% qui ont reçu une thérapie autologue par cellules CAR anti-CD19) après la randomisation.
Huit pour cent (8%) des patients dans le bras YESCARTA ont reçu ensuite une immunothérapie cellulaire hors protocole (y compris 7% qui ont reçu une thérapie autologue par cellules CAR anti-CD19 et y compris les 10 patients (6%) qui ont reçu un traitement répété par YESCARTA).
Greffe subséquente de cellules souches
Quatre pour cent (4%) des patients dans le bras de traitement standard et 7% dans le bras YESCARTA ont reçu ensuite un traitement à haute dose et une greffe autologue de cellules souches [HDT + GCSA], et 4% des patients dans le bras de traitement standard et 8% des patients dans le bras YESCARTA ont reçu ensuite une allogreffe de cellules souches (GCS).
Étude clinique (ZUMA-5), phase 2, LF récidivant ou réfractaire
L'efficacité et la sécurité de YESCARTA chez les patients adultes atteints de LF, ayant été traités par YESCARTA, ont été évaluées dans une étude de phase 2 multicentrique, en ouvert, monobras chez des patients atteints de LF récidivant ou réfractaire d'après la classification de l'OMS de 2016.
Les patients éligibles étaient âgés de ≥18 ans et présentaient une maladie récidivante ou réfractaire après au moins 2 lignes de traitement précédentes.
Le traitement précédent devait avoir inclus un anticorps monoclonal anti-CD20 associé à un agent alkylant (l'anticorps anti-CD20 en monothérapie ne comptait pas comme ligne de traitement). Les patients présentant une MS (sans récidive) > 1 an après la fin du dernier traitement étaient exclus. Les patients présentant un lymphome du SNC, ou ayant déjà fait l'objet d'une GCS allogénique ou ayant reçu un traitement antérieur par cellules CAR T anti-CD19 ou un autre traitement à base de cellules T génétiquement modifiées ont été exclus. Les patients présentant des antécédents de troubles du SNC (tels que convulsions ou ischémie vasculaire cérébrale), une fraction d'éjection cardiaque inférieure à 50%, ou une saturation en oxygène en air ambiant inférieure à 92%, ou une maladie auto-immune nécessitant un traitement immunosuppresseur systémique n'étaient pas éligibles. L'étude excluait les patients présentant des infections actives ou sévères, un lymphome transformé ou une localisation au niveau du SNC.
Au moment de l'analyse principale, un total de 127 patients atteints de LF avaient été inclus dans l'étude, y compris 80 patients qui avaient reçu au moins 3 lignes de traitement précédentes, et tous présentaient une leucaphérèse. Au cours de la période comprise entre la date de clôture des données de l'analyse principale et la date de clôture des données de l'analyse de suivi à 24 mois, aucun autre patient atteint de LF n'a été inclus ou traité par YESCARTA.
La durée réelle du suivi a été de 26,55 mois (intervalle: 0,3 à 44,3 mois, étude toujours en cours). Parmi les 127 patients atteints de FL recrutés, 80 patients avaient reçu au moins 3 lignes de traitement précédentes. Les données démographiques de la population ayant reçu au moins 3 lignes de traitement précédentes étaient conformes à celles de la population globale. Un résumé des données démographiques des patients est fourni dans le Tableau 6.
Tableau 6: Résumé des caractéristiques de la population pour les patients atteints de LF de l'étude ZUMA-5 (analyse à 24 mois)
|
Paramètres
|
Patients ayant eu une leucaphérèse ≥2 lignes de traitement précédentes
(N = 127)
|
Patients ayant eu une leucaphérèse ≥3 lignes de traitement précédentes
(N = 80)
| |
Âge (ans)
| |
Médiane (min, max)
|
60 (34, 79)
|
60.5 (34, 79)
| |
≥65
|
31%
|
33%
| |
Sexe masculin
|
59%
|
61%
| |
Origine ethnique
| |
Caucasien
|
92%
|
93%
| |
Asiatique
|
2%
|
4%
| |
Afro-américain
|
3%
|
3%
| |
Statut ECOG
| |
ECOG 0
|
62%
|
58%
| |
ECOG 1
|
38%
|
43%
| |
Forte masse tumorale, telle que définie par les critères GELF
|
51%
|
56%
| |
Nombre médian de traitements précédents (min; max)
|
3 (1, 10)
|
4 (3, 10)
| |
Patients atteints d'une maladie réfractaire à ≥2 lignes de traitement précédentes
|
29%
|
24%
| |
Patients atteints d'une maladie de stade III/IV
|
86%
|
88%
| |
Traitement antérieur par un inhibiteur de PI3K
|
28%
|
43%
| |
Délai de rechute < 24 mois après le premier traitement par association chimiothérapie et anti-CD20
|
56%
|
54%
|
ECOG, Eastern Cooperative Oncology Group; GELF, Groupe d'Étude des Lymphomes Folliculaires.
YESCARTA a été administré en une perfusion intraveineuse unique à une dose cible de 2 x 106 cellules CAR T anti-CD19/kg après une chimiothérapie lymphodéplétive par cyclophosphamide (500 mg/m² par voie intraveineuse) et fludarabine (30 mg/m2 par voie intraveineuse), tous deux administrés les 5e, 4e et 3e jours avant YESCARTA. Tous les patients ont été hospitalisés dans un but d'observation pendant au moins 7 jours après la perfusion de YESCARTA. L'administration et la surveillance de YESCARTA dans ZUMA-5 sont conformes à la procédure appliquée dans ZUMA-1.
L'analyse principale a été réalisée lorsque 84 patients atteints de LF recrutés consécutivement ont eu un suivi minimum de 12 mois à partir de la première évaluation de la réponse. Le critère d'évaluation principal était le dORR. Les critères d'évaluation secondaires comprenaient le taux de CR, le ORR et la CR chez les patients ayant reçu au moins 3 lignes de traitement précédentes, la DOR, l'OS, la survie sans progression (PFS) et l'incidence des effets indésirables. Trois des 127 patients atteints de LF inclus au moment de l'analyse principale n'ont pas été traités, principalement en raison du non-respect des critères d'inclusion ou du décès avant le traitement. Une analyse de suivi à 24 mois a été réalisée, lorsque 86 patients atteints de LF avaient un suivi minimum de 24 mois après la perfusion.
Lors de l'analyse de suivi à 24 mois, aucun patient supplémentaire n'a eu de leucaphérèse ni n'a été traité par YESCARTA. Le délai médian entre la leucaphérèse et la libération du produit était de 12 jours (intervalle: 10 à 37 jours), le délai médian entre la leucaphérèse et la réception du produit fini était de 17 jours (intervalle: 13 à 72 jours) et le délai médian entre la leucaphérèse et la perfusion de YESCARTA était de 27 jours (intervalle:19 à 330 jours). La dose médiane était de 2,0 × 106 cellules CAR T anti-CD19/kg.
Parmi les 127 patients atteints de LF inclus, 80 avaient reçu au moins 3 lignes de traitement précédentes. Le ORR était de 91% et le taux de CR était de 79%. Le délai médian de réponse était de 0,99 mois (intervalle: 0,8 à 3,1 mois), la DOR médiane était de 38,6 mois et la proportion de répondeurs toujours en réponse était de 55% jusqu'au mois 24. L'analyse des sous-groupes comprenait le ORR chez les patients réfractaires (89%) ainsi que les patients présentant un score FLIPI ≥3 (95%), une charge tumorale élevée (91%), une progression de la maladie dans les 24 mois suivant la première immunothérapie (91%) et un traitement antérieur par un inhibiteur de PI3K (91%). Les principaux résultats d'efficacité pour les patients atteints de LF ayant reçu au moins 3 lignes de traitement précédentes sont résumés dans le Tableau 7. Vingt-neuf des 80 patients atteints de LF qui avaient reçu au moins 3 lignes de traitement précédentes ont initialement obtenu une PR, dont 19 ont ensuite obtenu une CR.
Tableau 7: Résumé des résultats d'efficacité pour l'ensemble des patients atteints de LF inclus dans ZUMA-5 ayant reçu au moins 3 lignes de traitement précédentes (analyse à 24 mois)
|
Paramètres
|
Ensemble des patients ayant eu une leucaphérèse
N = 80
| |
ORRa, (%) [IC à 95%]
|
91% [83, 96]
| |
CR, (%) [IC à 95%]
|
79% [68, 87]
| |
PR, (%) [IC à 95%]
|
13% [6, 22]
| |
DORb, médiane en mois [IC à 95%]
|
38.6
| |
(intervalle)
|
(24.7, NE)
| |
DOR min, max (mois)
|
(0.0, 38,6)
| |
Réponse en cours (n)
|
44
| |
Taux de rémission continuec % (IC à 95%)
|
| |
12 mois
|
78,1 (66,2, 86,2)
| |
18 mois
|
72,8 (60,2, 82,0)
| |
24 mois
|
65,4 (51,2, 76,4)
| |
Survie sans progression, médiane, mois [IC à 95%]
|
40,2 (26,6, NE)
| |
Survie globale, médiane (mois) [IC à 95%]
|
NE (40,2, NE)
| |
Survie globale, % [IC à 95%]
|
| |
12 mois
|
96,2 (88,7, 98,8)
| |
18 mois
|
91,1 (82,2, 95,7)
| |
24 mois
|
85,2 (74,7, 91,5)
|
CR, réponse complète; DOR, durée de réponse; IC, intervalle de confiance; NE, non évaluable; dORR, taux de réponse objective; PR, réponse partielle.
a. Selon la classification de Lugano du groupe de travail international (Cheson 2014), évalué par le comité indépendant d'examen radiologique.
b. Parmi tous les répondeurs, la DOR est mesurée à partir de la date de la première réponse objective jusqu'à la date de la progression ou du décès.
c. Mesuré à partir de la date de la première réponse objective jusqu'à la date de la progression ou du décès.
PharmacocinétiqueCinétique cellulaire
Absorption
YESCARTA consiste en des cellules T autologues humaines. Les produits métaboliques attendus sont des produits de dégradation cellulaire typiques qui résultent des mécanismes normaux de clairance cellulaire. Les cellules CAR T perfusées devraient donc être éliminées avec le temps.
Après la perfusion de YESCARTA, les cellules CAR T anti-CD19 ont présenté une expansion initiale rapide suivie d'une diminution jusqu'à atteindre des taux proches des taux initiaux en 3 mois.
Les pics de concentration des cellules CAR T anti-CD19 survenaient dans les 7 à 14 premiers jours après la perfusion de YESCARTA. L'âge (intervalle: 21 à 80 ans) et le sexe n'ont eu aucun impact significatif sur l'ASC et les pics de concentration de YESCARTA.
Parmi les patients atteints de LDGCB dans ZUMA-1, le pic médian de concentration des cellules CAR T anti-CD19 dans le sang (Cmax) était de 38,3 cellules/µl (intervalle: 0,8-1513,7 cellules/μl); ce taux a diminué à 1 mois après la perfusion de YESCARTA jusqu'à une médiane de 2,1 cellules/µl (intervalle: 0-167,4 cellules/μl) et à une médiane de 0,4 cellules/µl (intervalle: 0-28,4 cellules/μl) à 3 mois après la perfusion de YESCARTA.
Parmi les patients dans ZUMA-7, le pic médian de concentration des cellules CAR T anti-CD19 dans le sang était de 25,84 cellules/µl (intervalle: 0,04 à 1173,25 cellules/μl); chez les patients évaluables ce taux a baissé jusqu'au taux initial au bout de 3 mois (0,35 cellule/µl; intervalle: 0,00 à 28,44 cellules/µl), mais chez 12 des 20 patients évaluables les cellules étaient encore détectables jusqu'à 24 mois après le traitement.
Parmi les patients atteints de LF, le pic médian des taux de cellules CAR T anti-CD19 dans le sang (Cmax) était de 39,4 cellules/µl (intervalle: 0,5-1415,4 cellules/μl). Le délai médian jusqu'au pic des cellules CAR T anti-CD19 dans le sang était de 8 jours après la perfusion (intervalle: 8 à 371 jours). Au bout de 3 mois, les taux de cellules CAR T anti-CD19 ont diminué jusqu'à atteindre des taux proches des valeurs enregistrées à l'inclusion, avec une médiane de 0.4 cellules/μl à 3 mois (intervalle: 0 à 15,8 cellules/μl).
Parmi les patients atteints de LDGCB et de LMPGB dans ZUMA-1, le nombre de cellules CAR T anti-CD19 dans le sang a été positivement corrélé à une réponse objective (CR ou PR). Les pics médians de concentration des cellules CAR T anti-CD19 chez les répondeurs (n = 73) étaient 205% plus élevées que les concentrations correspondantes chez les non-répondeurs (n = 23) (43,6 cellules/μl vs 21,2 cellules/μl). L'ASCjour 0-28 médiane chez les répondeurs (n = 73) était supérieure de 251% à la concentration correspondante chez les non-répondeurs (n = 23) (557,1 jours x cellules/μl vs 222,0 jours x cellules/μl). Le délai jusqu'à ce que le pic de concentration soit atteint était de 8 jours (7 jours après le jour de la perfusion de YESCARTA).
Les données recueillies pendant la période de suivi après la perfusion de YESCARTA ont montré qu'aucune cellule CAR T anti-CD19 génétiquement modifiée n'était détectable chez 13 patients sur 34 présentant une poursuite de la réponse après 24 mois alors qu'on détectait des valeurs très faibles de cellules CAR T anti-CD19 génétiquement modifiées chez 21 patients sur 34.
Certains patients ont eu besoin de tocilizumab et de corticostéroïdes pour le traitement de CRS et de neurotoxicités/d'ICANS. Les patients traités par tocilizumab (n=44) présentaient des taux de cellules CAR T anti-CD19 plus élevés de 262% et 232%, mesurés respectivement par l'ASCjour 0-28 et le pic de concentration, par rapport aux patients n'ayant pas reçu de tocilizumab (n=57). De même, les patients ayant reçu des corticoïdes (n=26) présentaient une ASCjour 0-28 et un pic de concentration plus élevés de 217% et de 155% que les patients n'ayant pas reçu de corticostéroïdes (n = 75).
Parmi les patients dans ZUMA-7, le nombre de cellules CAR T anti-CD19 dans le sang a été positivement corrélé à une réponse objective (RC ou RP). Les pics médians de concentration des cellules CAR T anti-CD19 chez les répondeurs (n = 142) étaient supérieurs de 275% environ aux concentrations correspondantes chez les non-répondeurs (n = 20) (28,9 cellules/μl contre 10,5 cellules/μl). L'ASCjour 0-28 médiane chez les patients répondeurs (n = 142) a été supérieure de 417% environ à la concentration correspondante chez les non-répondeurs (n = 20) (292,9 jours x cellules/μl contre 70,1 jours x cellules/μl). Le délai jusqu'à ce que soient atteints les pics de concentration était de 8 jours (7 jours après le jour de la perfusion de YESCARTA).
Les données recueillies pendant la période de suivi après la perfusion de YESCARTA ont montré qu'aucune cellule CAR T anti-CD19 génétiquement modifiée n'était détectable chez 18 patients sur 28 présentant une poursuite de la réponse et des taux évaluables de cellules CAR T après 24 mois alors qu'on détectait des valeurs très faibles de cellules CAR T génétiquement modifiées chez 10 patients sur 28. Certains patients ont eu besoin de tocilizumab et de corticostéroïdes pour le traitement de CRS et de toxicités neurologiques. Les patients traités par tocilizumab (n = 112) présentaient des taux de cellules CAR T anti-CD19 plus élevés de 215% et 274%, mesurés respectivement par l'ASCjour 0-28 et le pic de concentration, en comparaison des patients n'ayant pas reçu de tocilizumab (n = 58). De même, les patients ayant reçu des corticoïdes (n = 77) présentaient une ASCjour 0-28 et un pic de concentration plus élevés de 272% et de 176% que les patients n'ayant pas reçu de corticostéroïdes (n = 93).
Parmi les patients atteints de LF, le pic médian des taux de cellules CAR T anti-CD19 chez les répondeurs (n = 79) par rapport aux non-répondeurs (n = 3) était de 37,62 cellules/μl et 35,31 cellules/μl, respectivement. L'ASCjour 0-28 médiane chez les répondeurs par rapport aux non-répondeurs était de 451,17 cellules/μl•jours et 247,14 cellules/μl•jours, respectivement.
Distribution
Aucune donnée n'est disponible.
Métabolisme
Aucune donnée n'est disponible.
Élimination
YESCARTA est constitué de cellules T humaines autologues, on s'attend à ce que les produits de leur métabolisme soient des produits de dégradation cellulaires typiques, résultant de mécanismes de destruction cellulaire normaux. Il est donc attendu que les cellules CAR T perfusées soient détruites au fil du temps. Le nombre de cellules CAR T anti-CD19 a chuté au plus bas niveau jusqu'à 3 mois suivant la perfusion (intervalle: 0,000 à 28,410 cellules/μl dans ZUMA-1 et intervalle: 0,000 à 28,440 cellules/μl dans ZUMA-7). Aucune expansion secondaire des cellules CAR T n'a été observée dans ZUMA-1. Une expansion secondaire a été observée chez 2 patients dans ZUMA-5.
Les données recueillies pendant la période de suivi après la perfusion de YESCARTA ont montré qu'aucune cellule CAR T anti-CD19 génétiquement modifiée n'était détectable chez 13 patients sur 34 présentant une poursuite de la réponse après 24 mois alors qu'on détectait des valeurs très faibles de cellules CAR T anti-CD19 génétiquement modifiées chez 21 patients sur 34.
Cinétique pour certains groupes de patients
Âge et sexe
L'âge (intervalle: 21 à 81 ans) et le sexe n'avaient aucun effet significatif sur l'ASCjour 0-28 ni sur le pic de concentration de YESCARTA.
Troubles de la fonction hépatique
Aucune étude n'a été effectuée en cas de troubles de la fonction hépatique avec YESCARTA.
Troubles de la fonction rénale
Aucune étude n'a été effectuée en cas de troubles de la fonction rénale avec YESCARTA.
Données précliniquesYESCARTA contient des cellules T humaines génétiquement modifiées, qui réagissent particulièrement au CD19 d'origine humaine; par conséquent il n'existe pas de test représentatif in vitro, de modèles ex vivo ou de modèles in vivo, permettant d'analyser rigoureusement les caractéristiques toxicologiques de ce produit d'origine humaine. De ce fait, au cours du développement du médicament, aucune étude toxicologique traditionnelle n'a été réalisée.
Aucune étude de carcinogénicité ou de génotoxicité n'a été réalisée avec YESCARTA.
Aucune étude n'a été réalisée pour évaluer les effets de YESCARTA sur la fertilité, la reproduction et le développement.
Remarques particulièresIncompatibilités
Non pertinent.
Stabilité
YESCARTA est stable pendant 1 an quand il est conservé congelé dans la phase vapeur de l'azote liquide (≤ -150°C).
YESCARTA est stable à température ambiante (20°C à 25°C) jusqu'à 3 heures après décongélation complète. Afin de garantir que la stabilité est conservée après décongélation, la perfusion de YESCARTA doit commencer dans les 30 minutes suivant la fin de la décongélation complète et la durée totale de la perfusion ne doit pas dépasser 30 minutes (voir «Posologie/Mode d'emploi»).
Le médicament décongelé ne doit pas être recongelé.
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Les poches de YESCARTA doivent être conservées dans la phase vapeur de l'azote liquide (≤ -150°C) et elles doivent rester congelées jusqu'à ce que le patient soit prêt pour le traitement, afin de garantir que le patient reçoive des cellules autologues viables, capables de division cellulaire.
YESCARTA peut être conservé une seule fois à -80°C (±10°C) pendant une durée maximale de 90 jours. Dès lors que le produit est conservé à -80°C (±10°C), il doit être utilisé dans ce délai de 90 jours ou avant la date de péremption, selon ce qui survient en premier. Au-delà de ces dates, le médicament ne doit plus être utilisé et doit être éliminé.
Tenir hors de portée des enfants.
Pour les conditions de conservation du médicament après la décongélation, voir «Stabilité».
Précautions particulières concernant l'élimination et remarques concernant la manipulation
Une irradiation pourrait entraîner l'inactivation du médicament.
Ce médicament contient des cellules génétiquement modifiées. Les recommandations de biosécurité en vigueur localement pour ce médicament doivent être suivies pour les médicaments non utilisés ainsi que pour les déchets. Ce médicament peut avoir un pouvoir infectieux (p.ex. VIH, VHB ou VHC). Le personnel médical doit respecter les précautions de sécurité pour la manipulation et l'élimination du médicament. Les gants constituent un équipement de protection personnelle obligatoire pour l'administration de YESCARTA. Lors de la décontamination des surfaces et l'élimination du produit utilisé ayant été en contact avec YESCARTA, les recommandations de biosécurité en vigueur localement doivent être respectées.
Numéro d’autorisation66986 (Swissmedic)
PrésentationYESCARTA, une dispersion pour perfusion contenant 2 x 108 cellules/68 ml au maximum [A]
YESCARTA est livré en poche de cryoconservation en éthylène-acétate de vinyle.
Chaque poche de cryoconservation est emballée individuellement dans une cassette de transport.
Titulaire de l’autorisationGilead Sciences Switzerland Sàrl, Zug
Mise à jour de l’informationDécembre 2025
|