CompositionPrincipes actifs
Toxinum botulinicum A*.
* de Clostridium botulinum
Excipients
Albuminum humanum, 0,45 (50U) resp. 0,9 (100U) mg Natrii chloridum.
Un flacon de BOTOX 50 unités Allergan contient 0,18 mg de sodium.
Un flacon de BOTOX 100 unités Allergan contient 0,35 mg de sodium.
Indications/Possibilités d’emploiBOTOX est indiqué pour:
Affections neurologiques:
·traitement symptomatique du blépharospasme, de l'hémispasme facial et des dystonies focales associées, ainsi que pour la correction du strabisme chez les patients de plus de 12 ans.
·traitement symptomatique de la dystonie cervicale (torticolis spasmodique) chez l'adulte.
·traitement symptomatique de la spasticité focale des membres supérieurs et inférieurs chez l'adulte.
·traitement symptomatique de la spasticité focale des membres supérieurs et inférieurs chez les adolescents et les enfants à partir de 2 ans.
·prophylaxie des céphalées chez les patients adultes atteints de migraine chronique.
Affections vésicales:
·traitement de la vessie hyperactive accompagnée des symptômes suivants: incontinence urinaire, mictions impérieuses et mictions fréquentes, chez le patient adulte qui ne répond pas suffisamment aux anticholinergiques ou qui présente une intolérance à ces médicaments.
·traitement de l'incontinence urinaire causée par l'hyperactivité neurogène du détrusor associée à une affection neurologique (p.ex. lésion médullaire, sclérose en plaques) chez l'adulte.
·traitement de l'hyperactivité neurogène du détrusor associée à une affection neurologique (comme p.ex. le spina bifida, une lésion de la moelle épinière) chez les patients pédiatriques à partir de 5 ans, dont la vessie est vidangée de manière fiable par un sondage régulier à usage unique et qui ne répondent pas suffisamment aux médicaments anticholinergiques ou ne les tolèrent pas.
Affections de la peau et apparentées:
·traitement de l'hyperhidrose axillaire primaire chez l'adulte.
Posologie/Mode d’emploiLes unités de toxine botulique ne sont pas applicables à d'autres préparations. Les posologies exprimées en unités Allergan sont différentes de celles d'autres préparations de toxine botulique.
Voir «Remarques particulières» quant à «Remarques concernant la manipulation» et «Élimination» des flacons.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Instructions posologiques particulières
Patients âgés
Aucun ajustement spécifique de la posologie n'est nécessaire pour l'utilisation chez les patients âgés. Le dosage initial doit commencer à la dose minimale recommandée pour l'indication spécifique. Pour les injections répétées, il est recommandé d'administrer la plus faible dose efficace en respectant entre les injections l'intervalle le plus long possible qui est cliniquement indiqué. Les patients âgés présentant une anamnèse médicale importante et des médicaments concomitants doivent être traités avec précautions (pour des informations supplémentaires voir les rubriques «Effets indésirables» et «Propriétés/Effets»).
Enfants et adolescents
Il n'existe aucune étude sur la sécurité et l'efficacité du BOTOX dans le traitement du blépharospasme, du spasme hémifacial et du strabisme chez les enfants de moins de 12 ans.
La sécurité et l'efficacité du BOTOX dans le traitement de l'hyperactivité neurogène du détrusor chez les enfants de moins de 5 ans n'ont pas été étudiées.
La sécurité et l'efficacité de BOTOX pour le traitement de l'hyperactivité vésicale n'ont pas été démontrées chez les patients de moins de 18 ans (voir « Efficacité clinique» pour plus d'informations).
La sécurité et l'efficacité de BOTOX pour le traitement de la dystonie cervicale, la prophylaxie des céphalées en cas de migraine chronique et le traitement de l'hyperhidrose axillaire primaire n'ont pas été étudiées chez les enfants et les adolescents (âgés de moins de 18 ans).
Mode d'administration
BOTOX doit seulement être administré par des médecins possédant la qualification et les connaissances professionnelles adéquates dans le traitement et l'utilisation de l'équipement nécessaire. En fonction de l'indication, le diagnostic et l'administration de BOTOX doivent être effectués dans la mesure du possible en collaboration avec un neurologue, un ophtalmologiste, un pédiatre, un orthopédiste pédiatrique, un dermatologue ou un urologue.
On ne dispose pas de données cliniques suffisantes pour déterminer une posologie générale et un nombre fixe de sites d'injection dans chaque muscle. Le traitement doit donc être mis en place individuellement par le médecin spécialiste traitant le patient. Les doses optimales doivent être déterminées par titration. Le cas échéant, le potentiel de formation d'anticorps neutralisants peut être minimisé en administrant la dose efficace la plus faible aux intervalles les plus longs indiqués cliniquement entre les injections.
AFFECTIONS NEUROLOGIQUES:
Blépharospasme/hémispasme facial
Pour les patients de plus de 12 ans uniquement
Aiguille recommandée: Aiguille stérile de 27 à 30 gauges/0,40 à 0,30 mm.
Instruction d'administration: Le guidage électromyographique n'est pas nécessaire.
Dose recommandée: La dose initiale recommandée est de 1,25 à 2,5 unités, injectée dans les parties prétarsales médiale et latérale du muscle orbiculaire de l'œil de la paupière supérieure et dans la partie prétarsale latérale du muscle orbiculaire de l'œil de la paupière inférieure. Le produit peut également être injecté dans d'autres sites de l'arcade sourcilière, de la partie latérale du muscle orbiculaire de l'œil et de la zone faciale supérieure, si des spasmes gênent la vision.
Dose totale maximale: La dose initiale ne doit pas dépasser 25 unités par œil. Pour le traitement du blépharospasme la dose totale ne doit pas dépasser de 100 unités toutes les 12 semaines.
Informations complémentaires: Les injections à proximité du muscle releveur de la paupière supérieure sont à éviter pour éviter le risque de ptose. En raison de la diffusion de la solution de toxine botulique de type A dans le muscle oblique inférieur, une diplopie peut se manifester. Cet effet indésirable peut être réduit en évitant les injections médiales dans la paupière inférieure. Les sites d'injection possibles sont représentés ci-après:
Figure 1: Sites d'injection possibles

En général, l'effet initial des injections s'observe dans les trois jours suivants et atteint un maximum une à deux semaines après le traitement. L'effet de chaque traitement persiste environ 3 mois, après quoi les injections peuvent être répétées sans limitation. Lors des nouvelles séances de traitement, la dose peut être augmentée jusqu'à être doublée, si la réponse au traitement initial est jugée insuffisante.
Toutefois, l'injection de plus de 5,0 unités par site d'injection semble peu bénéfique. De même, un intervalle inférieur à trois mois entre deux traitements n'apporte aucun bénéfice supplémentaire.
Les patients atteints d'hémispasme facial ou de troubles du nerf facial (VII) doivent être traités comme pour un blépharospasme unilatéral et les autres muscles faciaux touchés doivent être traités par des injections de BOTOX en fonction du degré de spasme.
Strabisme
Pour les patients de plus de 12 ans uniquement
Aiguille recommandée: Aiguille stérile de 27 gauges/0,40 mm.
Instruction d'administration: Une solution de BOTOX contenant 2,5 unités par 0,1 ml (voir «Remarques concernant la manipulation» et «Élimination» sous «Remarques particulières»).
BOTOX est destiné à être injecté dans les muscles extra-oculaires en utilisant un guidage électromyographique.
Afin de préparer l'œil à l'injection de BOTOX, quelques gouttes d'un anesthésique local et d'une solution oculaire décongestionnante doivent être administrées quelques minutes avant l'injection.
Dose recommandée: Initialement, les doses les plus faibles doivent être utilisées pour le traitement des légères divergences et des doses plus élevées pour les divergences plus marquées.
Les doses initiales suivantes en unités sont recommandées (la dose la plus faible est prévue pour des angles de strabisme plus petits):
·pour les muscles verticaux et un strabisme horizontal de moins de 20 dioptries prismatiques: 1,25 unité à 2,5 unités (0,05 à 0,10 ml) dans chaque muscle.
·pour un strabisme horizontal de 20 à 50 dioptries prismatiques: 2,5 unités à 5,0 unités (0,10 à 0,20 ml) dans chaque muscle.
·pour une paralysie du nerf oculomoteur externe, qui perdure depuis un mois ou plus: 1,25 unité à 2,5 unités dans le muscle rectus medialis.
Dose totale maximale: La dose maximale recommandée pour une injection unique dans un muscle de l'œil est de 25 unités. Le volume d'injection recommandé de BOTOX à administrer pour le traitement du strabisme est de 0,05 à 0,15 ml par muscle.
Informations complémentaires: Les doses initiales de BOTOX dilué provoquent habituellement une paralysie des muscles injectés un à deux jours après l'injection. L'intensité de la paralyse augmente pendant la première semaine. Cette paralysie dure 2 à 6 semaines et se résorbe progressivement pendant une période à peu près équivalente. Les sur-corrections durant plus de 6 mois sont rares.
Environ la moitié des patients traités nécessitent des doses supplémentaires en raison d'une paralysie insuffisante du muscle après la dose initiale ou à cause de facteurs mécaniques tels que des divergences marquées ou une limitation ou en raison du manque de fusion binoculaire motrice afin de stabiliser l'adaptation.
Doses consécutives en cas de strabisme résiduel ou de récidive:
Il est recommandé de réexaminer les patients 7 à 14 jours après chaque injection pour évaluer l'effet de la dose. Les doses consécutives chez les patients présentant une paralysie suffisante du muscle cible doivent être identiques à la dose initiale.
Chez les patients présentant une paralysie incomplète du muscle cible, les doses consécutives peuvent aller jusqu'au double de la dose précédente. Il convient de n'effectuer aucune administration consécutive tant que l'effet de la dose précédente ne s'est pas résorbé, c'est-à-dire tant que les fonctions du muscle injecté et des muscles voisins ne sont pas rétablies.
Dystonie cervicale
Adultes seulement
Aiguille recommandée: Aiguille stérile de taille appropriée (généralement 25 à 30 gauges/0,50 à 0,30 mm).
Instruction d'administration: Dans les études cliniques sur le traitement de la dystonie cervicale, BOTOX a été habituellement injecté dans les muscles suivants: sterno-cléido-mastoïdien, élévateur de l'omoplate, scalène, splénius de la tête, semi-épineux, longissimus et/ou trapèze. Cette liste n'est pas exhaustive, car tout muscle qui contrôle la position de la tête peut être atteint et dès lors nécessiter un traitement.
Chez les femmes, on observe plus fréquemment des effets secondaires dose-dépendants. Il convient donc de prendre en compte la masse musculaire et le degré d'hypertrophie ou d'atrophie du muscle à traiter lors de la sélection de la dose appropriée. Le schéma d'activation musculaire de la dystonie cervicale peut changer spontanément sans que le tableau clinique de la dystonie en soit modifié.
En cas de difficulté pour identifier clairement le muscle touché, les injections doivent être effectuées avec une assistance électromyographique.
Dose recommandée: Lors de la première séance de traitement, la dose totale injectée ne doit pas dépasser 200 unités. Lors des séances suivantes, la dose sera ajustée en fonction de la réponse initiale.
Dans les premières études cliniques contrôlées visant à prouver l'innocuité et l'efficacité du produit pour le traitement de la dystonie cervicale, les doses de BOTOX reconstitué allaient de 140 à 280 unités. Dans des études plus récentes, les doses allaient de 95 à 360 unités (avec une moyenne de 240 unités environ).
L'administration initiale chez un patient jamais traité auparavant doit commencer à la dose minimale efficace. Il ne faut pas administrer plus de 50 unités dans un même site. Le sterno-cléido-mastoïdien ne doit pas recevoir plus de 100 unités. Afin de minimiser le risque de dysphagie, les injections dans le sterno-cléido-mastoïdien ne doivent pas être bilatérales.
Dose totale maximale: La dose totale de 300 unités par traitement ne doit pas être dépassée. Le nombre optimal de sites d'injection dépend de la taille du muscle. Ne pas répéter les injections à moins de 10 semaines d'intervalle.
Informations complémentaires: Les améliorations cliniques surviennent généralement dans les deux premières semaines après l'injection. Les effets cliniques maximaux apparaissent en général vers la sixième semaine après l'injection. La durée de l'effet rapportée dans les essais cliniques était très inégale (de 2 à 33 semaines), avec une durée moyenne d'environ 12 semaines.
Spasticité focale des membres supérieurs
Adultes
Aiguille recommandée: Une aiguille stérile de 25, 27 ou 30 gauges dans les muscles superficiels et une aiguille de 22 gauges dans les muscles plus profonds.
Instruction d'administration: Pour localiser les muscles concernés, le recours à un guidage électromyographique ou à la stimulation nerveuse peut être utile. Un contact plus uniforme avec les régions d'innervation du muscle peut être obtenu par l'administration de BOTOX à plusieurs sites d'injection. Cela peut être utile en particulier chez les muscles grands.
Dose recommandée: La posologie exacte doit être déterminée individuellement. La dose et le nombre des sites d'injection dépendent de la grosseur, du nombre et de l'emplacement des muscles impliqués, de la présence d'une faiblesse musculaire locale et de la réponse au traitement antérieur chez le patient.
De fortes doses peuvent entraîner une réduction plus durable de la tonicité musculaire. La sévérité de la spasticité musculaire et les groupes musculaires impliqués peuvent varier au cours du temps et il peut être nécessaire d'envisager une modification de la dose de BOTOX et des muscles à traiter.
Les doses initiales et les sites d'injection suivants sont recommandés:
|
Muscle
|
Posologie totale;
nombre de sites d'injection
| |
Biceps brachii
|
50 à 200 unités; jusqu'à 4 sites
| |
Flexor digitorum profundus
|
7,5 à 30 unités; 1 à 2 sites
| |
Flexor digitorum sublimis
|
7,5 à 30 unités; 1 à 2 sites
| |
Flexor carpi radialis
|
15 à 60 unités; 1 à 2 sites
| |
Flexor carpi ulnaris
|
10 à 40 unités; 1 à 2 sites
|
Dose totale maximale: Dans les études cliniques, la dose de 360 unités chez l'adulte n'a pas été dépassée. La dose totale a été répartie entre les muscles sélectionnés (habituellement, les muscles fléchisseurs du coude, du poignet et des doigts). Il convient généralement de ne pas dépasser une dose de 6 unités/kg de poids corporel.
Informations complémentaires: Habituellement, on obtient une amélioration du tonus musculaire en l'espace de 2 semaines, avec un effet maximum 4 à 6 semaines après le traitement. Des doses répétées ne doivent être administrées qu'une fois l'effet clinique de l'injection précédente a disparu, mais avec un intervalle de deux mois au moins.
Adolescents et enfants à partir de 2 ans
Aiguille recommandée: Taille d'aiguille stérile appropriée. La longueur de l'aiguille doit être choisie en fonction de la localisation et de la profondeur du muscle.
Instructions d'administration: Le guidage par EMG, la stimulation nerveuse ou l'échographie sont recommandés pour la localisation des muscles impliqués.
L'illustration ci-après Figure 2 présente les sites d'injection pour le traitement de la spasticité des membres supérieurs chez les enfants:
Figure 2: Sites d'injection pour le traitement de la spasticité des membres supérieurs chez les enfants
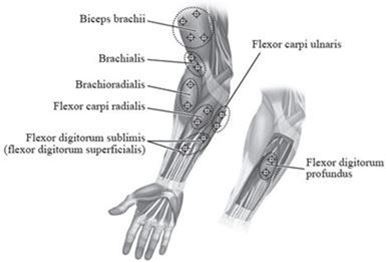
Dose recommandée: La dose recommandée pour le traitement de la spasticité des membres supérieurs chez l'enfant est de 3 à 6 unités/kg de poids corporel réparties entre les muscles affectés.
La posologie et les sites d'application suivants sont recommandés:
|
Muscle
|
Dose totale; nombre de sites d'injection
| |
Biceps brachii
|
1,5 à 3 unités /kg; réparties sur 4 sites
| |
Brachialis
|
1 à 2 unités/kg; réparties sur 2 sites
| |
Brachioradialis
|
0,5 à 1 unité/kg; répartie sur 2 sites
| |
Flexor carpi radialis
|
1 à 2 unités/kg; réparties sur 2 sites
| |
Flexor carpi ulnaris
|
1 à 2 unités/kg; réparties sur 2 sites
| |
Flexor digitorum profundus
|
0,5 à 1 unité/kg; répartie sur 2 sites
| |
Flexor digitorum sublimis
|
0,5 à 1 unité/kg; répartie sur 2 sites
|
Dose totale maximale: La dose totale de BOTOX par traitement des membres supérieurs ne doit pas dépasser 6 unités/kg de poids corporel ou 200 unités, selon la valeur la plus faible. Si le médecin traitant le juge approprié, un renouvellement du traitement doit être envisagé chez le patient lorsque l'effet clinique de l'injection précédente a diminué, mais pas avant 12 semaines après l'injection précédente. Lors du traitement simultané des membres supérieurs et inférieurs, la dose totale dans un intervalle de 12 semaines ne doit pas dépasser la limite inférieure de 10 unités/kg de poids corporel ou 340 unités.
Informations complémentaires: En règle générale, l'amélioration du tonus musculaire se produit dans les 7 jours suivant le traitement. Le traitement au BOTOX ne remplace pas les traitements de rééducation standard habituels.
Spasticité focale des membres inférieurs
Adultes
Aiguille recommandée: Aiguille stérile de calibre 25, 27 ou 30. La longueur de l'aiguille doit être choisie en fonction de la localisation et de la profondeur du muscle.
Instructions d'administration: Le guidage par EMG, la stimulation nerveuse ou l'échographie peuvent être utiles pour localiser les muscles concernés. L'application de BOTOX sur plusieurs sites d'injection permet d'obtenir un contact plus uniforme avec les zones d'innervation des muscles, ce qui est particulièrement utile pour les gros muscles.
L'illustration ci-après Figure 3 présente les sites d'injection pour la spasticité des membres inférieurs chez les adultes:
Figure 3: Sites d'injection pour la spasticité des membres inférieurs chez les adultes
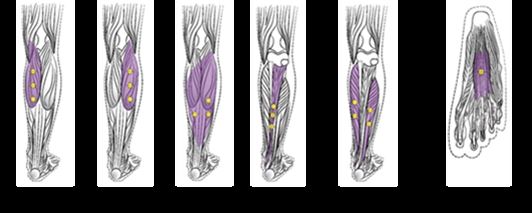
Dose recommandée: La dose recommandée pour le traitement des membres inférieurs, y compris la cheville et le pied, est de 300 à 400 unités réparties entre les muscles concernés énumérés dans le tableau ci-dessous. La dose totale maximale par traitement est de 400 unités. Le degré et le profil de la spasticité musculaire au moment de la réinjection peuvent nécessiter des modifications de la dose de BOTOX et des muscles à injecter.
La posologie et les sites d'application suivants sont recommandés:
|
Muscle
|
Dose totale; nombre de sites d'injection
| |
Chef médial du gastrocnémien
|
75 unités; 3 sites
| |
Chef latéral du gastrocnémien
|
75 unités, 3 sites
| |
Soleus
|
75 unités; 3 sites
| |
Tibialis posterior
|
75 unités; 3 sites
| |
Flexor hallucis longus
|
50 unités; 2 sites
| |
Flexor digitorum longus
|
50 unités; 2 sites
| |
Flexor digitorum brevis
|
25 unités; 1 site
|
Informations complémentaires: Le médecin traitant est libre d'envisager une réinjection si l'effet clinique de l'injection précédente s'estompe, mais pas avant 12 semaines après l'injection précédente.
Enfants à partir de 2 ans
Aiguille recommandée: Taille d'aiguille stérile appropriée. La longueur de l'aiguille doit être choisie en fonction de la localisation et de la profondeur du muscle.
Instructions d'administration: Le guidage par EMG, la stimulation nerveuse ou l'échographie sont recommandés pour la localisation des muscles impliqués.
L'illustration ci-après Figure 4 présente les sites d'injection pour le traitement de la spasticité des membres inférieurs chez les enfants:
Figure 4: Sites d'injection pour le traitement de la spasticité des membres inférieurs chez les enfants
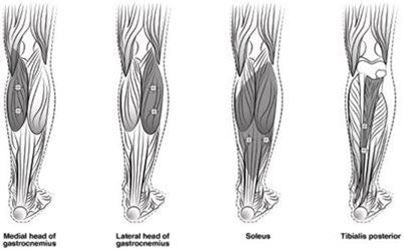
Dose recommandée: La dose recommandée pour le traitement de la spasticité des membres inférieurs chez l'enfant est de 4 à 8 unités/kg de poids corporel réparties entre les muscles affectés.
La posologie et les sites d'application suivants sont recommandés:
|
Muscle
|
Dose totale; nombre de sites d'injection
| |
Chef médial du gastrocnémien
|
1 à 2 unités/kg; réparties sur 2 sites
| |
Chef latéral du gastrocnémien
|
1 à 2 unités/kg; réparties sur 2 sites
| |
Soleus
|
1 à 2 unités/kg; réparties sur 2 sites
| |
Tibialis posterior
|
1 à 2 unités/kg; réparties sur 2 sites
|
Dose totale maximale: La dose totale de BOTOX par traitement des membres inférieurs ne doit pas dépasser 8 unités/kg de poids corporel ou 300 unités, selon la valeur la plus faible. Le médecin traitant est libre d'envisager une réinjection si l'effet clinique de l'injection précédente s'estompe, mais pas avant 12 semaines après l'injection précédente. Lors du traitement simultané des membres supérieurs et inférieurs, la dose totale dans un intervalle de 12 semaines ne doit pas dépasser la limite inférieure de 10 unités/kg de poids corporel ou 340 unités.
Informations complémentaires: En règle générale, l'amélioration du tonus musculaire se produit dans les 7 jours suivant le traitement. Le traitement au BOTOX ne remplace pas les traitements de rééducation standard habituels.
Prophylaxie des céphalées chez les patients adultes atteints de migraine chronique
Aiguille recommandée: Une aiguille de 30 gauges/13 mm est utilisée.
Instructions d'administration: Les injections doivent être réparties sur sept zones musculaires spécifiques de la tête et de la nuque, comme décrit dans le tableau ci-dessous. À l'exception du muscle procérus, qui doit être injecté sur un site (ligne médiane), tous les muscles doivent être injectés bilatéralement avec la dose minimale par muscle indiquée ci-après, la moitié des sites d'injection se situant donc du côté gauche et l'autre moitié du côté droit de la tête et de la nuque. Si un ou plusieurs points douloureux prédominent, des injections supplémentaires peuvent être administrées unilatéralement ou bilatéralement dans un maximum de 3 groupes musculaires spécifiques (occipital, temporal et trapèze), jusqu'à la dose maximale par muscle indiquée dans le tableau ci-dessous.
Le calendrier recommandé pour le renouvellement du traitement est de 12 semaines.
Les points d'injection sont représentés dans les illustrations ci-dessous:
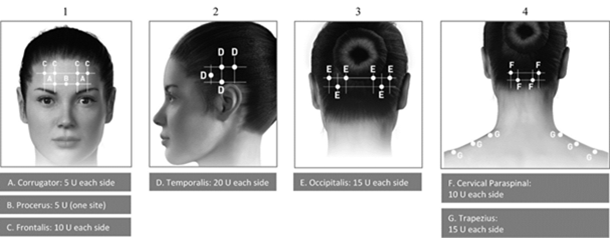
Les groupes de muscles recommandés pour les injections supplémentaires facultatives sont représentés dans les illustrations ci-dessous:
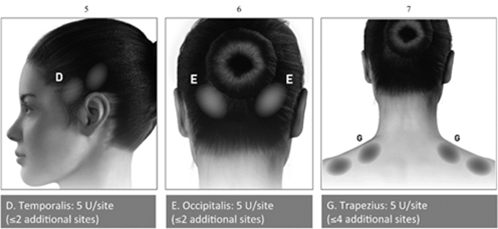
Dose recommandée: La dose recommandée est comprise entre 155 et 195 unités administrées par voie intramusculaire (i.m.) sous forme d'injections de 0,1 ml (5 unités) chacune, pratiquées dans 31 à 39 sites.
BOTOX - Posologie selon le muscle en cas de migraine chronique:
|
|
Dose recommandée
| |
Zone de la tête et de la nuque
|
Dose totale (nombre de points d'injectiona)
| |
Corrugatorb
|
10 unités (2 sites)
| |
Procérus
|
5 unités (1 site)
| |
Frontalb
|
20 unités (4 sites)
| |
Temporalb
|
40 unités (8 sites) jusqu'à 50 unités (jusqu'à 10 sites)
| |
Occipitalb
|
30 unités (6 sites) jusqu'à 40 unités (jusqu'à 8 sites)
| |
Groupe musculaire cervical paraspinalb
|
20 unités (4 sites)
| |
Trapèzeb
|
30 unités (6 sites) jusqu'à 50 unités (jusqu'à 10 sites)
| |
Plage de dose totale:
|
155 unités jusqu'à 195 unités
31 à 39 sites
|
a Chaque point d'injection i.m. = 0,1 ml = 5 unités BOTOX
b Dose répartie des deux côtés
Informations complémentaires: L'amélioration survient généralement au cours des 3 premières semaines après l'injection. En règle générale, l'effet persiste pendant 12 semaines après l'injection.
AFFECTIONS VÉSICALES:
Injection dans le muscle détrusor - adultes
Généralités
Instruction d'administration: Au moment du traitement, les patients ne doivent pas être atteints d'une infection urinaire aiguë.
Un traitement antibiotique prophylactique doit être administré 1 à 3 jours avant le traitement, le jour du traitement et 1 à 3 jours après le traitement.
Il est conseillé aux patients de cesser le traitement anti-agrégation plaquettaire au moins 3 jours avant l'injection. Les patients sous traitement anticoagulant doivent être pris en charge de façon appropriée pour réduire le risque d'hémorragie.
Hyperactivité vésicale – adultes
Aiguille recommandée: Un cystoscope flexible ou rigide peut être utilisé. Avant de commencer les injections, il faut remplir (préparer) l'aiguille d'injection stérile par environ 1 ml de BOTOX reconstitué (selon la longueur de l'aiguille) pour en retirer totalement l'air.
Instruction d'administration: Une instillation intravésicale avec un anesthésique local dilué avec ou sans sédation peut être utilisée avant l'injection, selon les pratiques locales. Si une instillation d'anesthésique locale est effectuée, la vessie doit être vidangée et rincée avec une solution de chlorure de sodium stérile avant les étapes suivantes du processus d'injection.
La solution de BOTOX reconstituée (100 unités /10 ml) est injectée dans le muscle détrusor au moyen d'un cystoscope flexible ou rigide en évitant le trigone. La vessie doit être instillée avec suffisamment de solution de chlorure de sodium afin d'obtenir une visualisation suffisante pour l'injection. Une dilatation exagérée doit cependant être évitée.
L'aiguille doit être introduite sur environ 2 mm dans le muscle détrusor et 20 injections de 0,5 ml (volume total de 10 ml) doivent être effectuées à une distance d'environ 1 cm (voir Figure 5).Comme injection finale, environ 1 ml de solution de chlorure de sodium physiologique stérile sera injecté afin que la dose totale soit administrée. Après administration des injections, la solution de chlorure de sodium utilisée pour la visualisation de la paroi vésicale ne sera pas drainée afin que le patient puisse démontrer sa capacité d'uriner avant sa sortie de la clinique. Le patient sera surveillé pendant au moins 30 minutes après l'injection et jusqu'à ce qu'une vidange spontanée de la vessie ait eu lieu.
Figure 5: Sites d'injection pour le traitement des troubles fonctionnels de la vessie
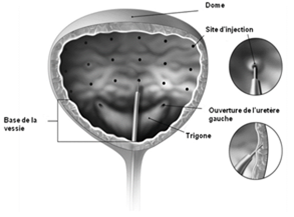
Dose recommandée: La dose recommandée est de 100 unités de BOTOX sous forme d'injections de 0,5 ml (5 unités), réparties sur 20 endroits du muscle détrusor.
Informations complémentaires: Une amélioration clinique peut se manifester dans un délai de 2 semaines. Une nouvelle injection devrait être envisagée pour les patients chez lesquels l'effet clinique de l'injection précédente a diminué (la durée médiane dans les études cliniques de phase 3 était de 166 jours [~24 semaines]). La nouvelle injection ne devrait cependant pas être effectuée dans les 3 mois qui suivent l'injection vésicale précédente.
Incontinence urinaire causée par l'hyperactivité neurogène du détrusor - adultes
Aiguille recommandée: Un cystoscope flexible ou rigide peut être utilisé. Avant de commencer les injections, il faut remplir (préparer) l'aiguille d'injection par environ 1 ml (selon la longueur de l'aiguille) pour en retirer totalement l'air.
Instruction d'administration: Il est possible d'instiller par voie intravésicale un anesthésique dilué, avec ou sans sédation, ou recourir à une anesthésie générale avant l'injection, selon les pratiques locales. Si un anesthésique local est instillé, la vessie doit être drainée et rincée à l'aide d'une solution saline stérile avant de poursuivre la procédure d'injection.
La solution reconstituée de BOTOX (200 unités /30 ml) est injectée dans le détrusor à l'aide d'un cystoscope flexible ou rigide, en évitant le trigone. Il faut instiller dans la vessie une quantité suffisante de solution saline pour une bonne visualisation des injections. Une dilatation exagérée doit cependant être évitée.
L'aiguille doit être introduite sur environ 2 mm à l'intérieur du détrusor et 30 injections de 1 ml chacune (volume total de 30 ml) doivent être effectuées avec un espace d'environ 1 cm (voir la figure ci-dessus). Pour la dernière injection, environ 1 ml de solution physiologique stérile doit être injecté pour que la dose complète puisse être administrée. Une fois les injections réalisées, la solution saline injectée dans la vessie pour permettre d'en voir les parois doit être drainée. Le patient doit être gardé en observation pendant au moins 30 minutes après l'intervention.
Dose recommandée: La dose recommandée est de 200 unités de BOTOX réparties en 30 injections d'un volume de 1 ml (~6,7 unités) dans le détrusor.
Informations complémentaires: Une amélioration clinique se produit en général dans les 2 semaines suivant l'intervention. De nouvelles injections sont envisageables chez le patient lorsque l'effet clinique des injections précédentes a diminué (la durée médiane dans les essais cliniques de phase 3 était de 256 à 295 jours (~36-42 semaines) pour BOTOX (à raison de 200 unités)), mais ces nouvelles injections ne doivent pas être faites moins de 3 mois après la dernière injection vésicale.
Hyperactivité neurogène du détrusor - patients pédiatriques âgés de 5 ans et plus
Généralités
Instruction d'administration: Les patients ne doivent pas souffrir d'infection urinaire aiguë (urinary tract infection, UTI) au moment du traitement. Des antibiotiques par voie orale doivent être administré à titre prophylactique 1 à 3 jours avant le traitement, le jour du traitement et 1 à 3 jours après le traitement, afin de réduire le risque de survenue d'une infection urinaire liée à l'intervention. Il est conseillé aux patients de cesser le traitement antiagrégant plaquettaire au moins 3 jours avant l'injection.
Alternativement, chez les patients recevant une anesthésie générale (ou une sédation consciente) pour le traitement de l'hyperactivité du détrusor liée à une maladie neurologique, une dose d'antibiotiques prophylactiques peut être administrée par voie IV avant le traitement, le jour du traitement. Généralement, l'utilisation d'aminoglycosides n'est pas recommandée (voir «Interactions»).
Les patients traités par antiagrégants plaquettaires doivent arrêter leur traitement au moins 3 jours avant l'injection. Les patients qui reçoivent un traitement anticoagulant doivent être pris en charge de façon appropriée pour réduire le risque d'hémorragie.
Lors de la réalisation d'une cystoscopie, il convient de prendre les précautions adéquates.
·Chez les patients âgés de 5 à moins de 12 ans: Avant l'injection, il convient d'envisager une anesthésie générale (ou une sédation consciente), conformément à la procédure habituelle locale.
·Chez les patients âgés de 12 ans et plus: Une instillation intra-vésicale d'un anesthésique local dilué, avec ou sans sédation ou une anesthésie générale doivent être envisagée avant le traitement par injections de BOTOX, conformément à la procédure habituelle locale.
Pour tous les groupes d'âge, il convient au moins d'envisager l'instillation intra-vésicale d'un anesthésique local dilué. Si une instillation d'anesthésique locale est réalisée, la vessie doit être vidangée et rincée avec une solution de chlorure de sodium stérile avant les étapes suivantes de la procédure d'injection.
Aiguille recommandée: Il est possible d'utiliser un cystoscope flexible ou rigide. L'aiguille d'injection stérile doit être remplie (amorcée) avec environ 1 ml de solution de dosage de BOTOX (en fonction de la longueur de l'aiguille) avant le début des injections, afin d'éliminer la totalité de l'air.
Instruction d'administration: Le BOTOX reconstitué est injecté dans le muscle détrusor à l'aide d'un cystoscope flexible ou rigide, en évitant le trigone. Il convient de remplir la vessie avec suffisamment de solution de chlorure de sodium afin d'obtenir une visualisation suffisante pour les injections, tout en évitant une distension excessive.
L'aiguille doit être introduite sur environ 2 mm dans le muscle détrusor et 20 injections de 0,5 ml chacune (volume total: 10 ml) doivent être réalisées avec une espace d'environ 1 cm (voir Figure 5). Comme injection finale, environ 1 ml de solution de chlorure de sodium physiologique stérile doit être injecté pour que le reste de BOTOX passe de l'aiguille au muscle détrusor et que la dose totale soit administrée. Après administration de toutes les injections, la solution de chlorure de sodium utilisée pour la visualisation de la paroi vésicale doit être vidée. Le patient doit être surveillé pendant encore au moins 30 minutes après la fin des injections.
Dose recommandée: Si le poids corporel du patient est supérieur ou égal à 34 kg, la dose recommandée s'élève à 200 unités de BOTOX par traitement, administrées sous forme d'injections intra-détrusor après dilution.
·Reconstituez le BOTOX pour que 20 unités de BOTOX par ml se trouvent dans le(s) flacon(s) de prélèvement:
·Flacons de prélèvement BOTOX avec 100 unités: Ajoutez 5 ml de solution de chlorure de sodium à 0,9 % sans conservateur et secouez le(s) flacon(s) de prélèvement avec précaution.
·Aspirez 10 ml du ou des flacons dans une seringue de dosage de 10 ml.
·Utilisez la solution immédiatement après la reconstitution dans la seringue. Éliminez la solution de chlorure de sodium à 0,9 % sans conservateur non utilisée.
Si le poids corporel du patient est inférieur à 34 kg, la dose recommandée s'élève à 6 unités de BOTOX par kg de poids et par traitement, administrées sous forme d'injection dans le détrusor:
·Reconstituez le BOTOX pour que 20 unités de BOTOX par ml se trouvent dans le(s) flacon(s) de prélèvement:
·Flacon(s) de prélèvement BOTOX avec 100 unités: Ajoutez 5 ml de solution de chlorure de sodium à 0,9 % sans conservateur à un flacon de prélèvement de 100 unités de BOTOX (lorsque la dose finale est inférieure ou égale à 100 U) ou à chacune de deux flacons de BOTOX de 100 unités (lorsque la dose finale est supérieure à 100 U) et secouez le/les flacon(s) de prélèvement avec précaution.
·Selon le poids corporel du patient, le BOTOX reconstitué à une concentration de 20 unités pour 1 ml doit être dilué davantage. Les instructions complémentaires concernant la dilution en fonction du poids du patient se rapportent à la quantité de BOTOX reconstitué (20 unités pour 1 ml) et à la solution de chlorure de sodium à 0,9 % sans conservateur supplémentaire, qui doivent être aspirés ensemble dans une seringue de dosage de 10 ml. Elles sont présentées dans le tableau 1:.
Tableau 1: Instructions de dilution du BOTOX et dose finale pour les patients ayant un poids corporel < 34 kg
|
Poids corporel
(kg)
|
Volume de BOTOX reconstitué (20 U/ml) et des diluants* (ml) à prélever dans la seringue de dosage, pour atteindre un volume final de 10 ml pour l'injection dans le muscle détrusor
|
Dose finale de BOTOX dans la seringue de dosage
| |
BOTOX
(ml)
|
Diluant*
(ml)
| |
12 à moins de 14
|
3,6
|
6,4
|
72 unités
| |
14 à moins de 16
|
4,2
|
5,8
|
84 unités
| |
16 à moins de 18
|
4,8
|
5,2
|
96 unités
| |
18 à moins de 20
|
5,4
|
4,6
|
108 unités
| |
20 à moins de 22
|
6
|
4
|
120 unités
| |
22 à moins de 24
|
6,6
|
3,4
|
132 unités
| |
24 à moins de 26
|
7,2
|
2,8
|
144 unités
| |
26 à moins de 28
|
7,8
|
2,2
|
156 unités
| |
28 à moins de 30
|
8,4
|
1,6
|
168 unités
| |
30 à moins de 32
|
9
|
1
|
180 unités
| |
32 à moins de 34
|
9,6
|
0,4
|
192 unités
|
* Solution de chlorure de sodium à 0,9 % sans conservateur
·Utilisez le BOTOX immédiatement après la reconstitution et la dilution dans la seringue. Éliminez la solution de chlorure de sodium à 0,9 % sans conservateur non utilisée.
Informations complémentaires: Une amélioration clinique survient généralement en l'espace de 2 semaines. On peut envisager de pratiquer une nouvelle injection chez les patients lorsque l'effet clinique de l'injection précédente diminue (la durée médiane, dans l'étude clinique en double aveugle à groupes parallèles, s'élevait à 207 jours [~30 semaines] à raison de 200 unités de BOTOX, adaptées en fonction du poids afin de ne pas dépasser 6 U/kg), en respectant toutefois un délai minimum de 3 mois après l'injection vésicale précédente.
AFFECTIONS DE LA PEAU ET APPARENTÉES:
Hyperhidrose axillaire primaire
Adultes seulement
Aiguille recommandée: Aiguille stérile de 30 gauges.
Instruction d'administration: Il est possible de délimiter la zone hyperhidrotique en utilisant des techniques de coloration standard, p.ex. le test iode-amidon de Minor.
Dose recommandée: 50 unités de BOTOX (100 unités /4,0 ml) sont injectés par voie intradermique, réparties équitablement entre plusieurs sites distants d'environ 1 à 2 cm, dans la zone hyperhidrotique de chaque aisselle atteinte.
Dose totale maximale: D'autres doses que 50 unités par aisselle n'ayant pas fait l'objet d'études, elles ne peuvent pas être recommandées. Les injections ne doivent pas être répétées plus fréquemment que toutes les 16 semaines.
Informations complémentaires: L'amélioration clinique se manifeste généralement dans la première semaine suivant l'injection. Des injections consécutives peuvent être effectuées lorsque les effets cliniques des injections précédentes diminuent et que le médecin traitant l'estime nécessaire. Par expérience, la réponse au traitement persiste pendant 4 à 7 mois.
TOUTES LES INDICATIONS:
En cas d'absence, un mois après la première injection, d'effet thérapeutique, il y à lieu de prendre les mesures suivantes:
·Vérification clinique, pouvant inclure un examen électromyographique dans un centre spécialisé, de l'action de la toxine sur le(s) muscle(s) traité(s).
·Analyse des causes de l'échec thérapeutique, p.ex. mauvaise sélection des muscles à traiter, dose insuffisante, technique d'injection inadaptée, apparition d'une contracture fixée, muscles antagonistes trop faibles, formation d'anticorps.
·Réévaluation de la pertinence du traitement par la toxine botulique de type A.
En l'absence de tout effet indésirable à l'issue de la première séance de traitement, mise en œuvre d'une deuxième séance en respectant les conditions suivantes:
1.ajustement de la dose en fonction de l'analyse de l'échec thérapeutique précédent,
2.utilisation du guidage électromyographique,
3.maintien d'un intervalle de trois mois entre deux séances.
En cas d'absence d'effet thérapeutique ou de diminution des effets après des injections répétées, d'autres méthodes thérapeutiques devront être envisagées.
Dans le traitement des adultes, y compris le traitement d'indications multiples, la dose maximale cumulée sur une période de 12 semaines ne doit pas dépasser 400 unités.
Dans le traitement des enfants, y compris le traitement d'indications multiples, la dose maximale cumulée de 10 unités/kg de poids corporel ou de 340 unités sur une période de 12 semaines, selon la plus faible de ces deux valeurs, ne doit pas être dépassée.
Contre-indications·Hypersensibilité connue à la substance active, à savoir la toxine botulique de type A, ou à l'un des excipients selon la composition.
·Présence d'une infection au niveau du (des) site(s) d'injection prévu(s).
Les injections de BOTOX dans le muscle détrusor sont également contre-indiquées:
·Chez les patients qui souffrent d'une infection urinaire au moment du traitement.
·Chez les patients qui présentent une rétention urinaire aiguë au moment du traitement et qui n'utilisent pas régulièrement un sondage vésical.
·Chez les patients qui ne veulent pas et/ou ne peuvent pas, en cas de besoin, recourir au sondage après le traitement.
Mises en garde et précautionsLes posologies recommandées et la fréquence d'administration de BOTOX ne doivent pas être dépassées, sous peine de causer un surdosage, une augmentation de la faiblesse musculaire, une diffusion de la toxine à distance du site d'administration et la formation d'anticorps neutralisants. La dose initiale chez des patients non traités auparavant doit être la dose recommandée la plus faible pour l'indication correspondante.
Les médecins et les patients doivent être conscients que des effets indésirables peuvent survenir même si les injections précédentes ont été bien tolérées. Il convient donc de prendre des précautions à chaque administration.
Des effets indésirables liés à la diffusion de la toxine à distance du site d'administration ont été rapportés (voir «Effets indésirables»). Dans quelques cas, ces effets ont été fatals et parfois associés à une dysphagie, une pneumonie et/ou des états de faiblesse. Des symptômes correspondent au mécanisme d'action de la toxine botulique et ont été rapportés plusieurs heures à plusieurs semaines après l'injection. Le risque d'apparition des symptômes est probablement élevé chez les patients souffrant de maladies susceptibles d'augmenter la prédisposition envers ces symptômes. Parmi ces patients, on trouve également des enfants et des adultes traités par de fortes doses pour cause de spasticité.
Le médecin traitant doit être familiarisé avec la technique électromyographique lorsqu'il injecte BOTOX pour traiter un strabisme. L'effet de BOTOX n'est pas assuré en présence d'écart supérieur à 50 dioptries en cas de strabisme de type restrictif, de syndrome de Duane ou de strabisme secondaire résultant d'une sur-correction chirurgicale antérieure du muscle antagoniste. Plusieurs cycles de traitement peuvent éventuellement être nécessaires.
Chez les patients traités par des doses thérapeutiques, une faiblesse musculaire accrue peut également survenir.
Avant le début du traitement par BOTOX, les bénéfices et les risques individuels doivent être évalués.
Des cas de dysphagie après des injections qui n'avaient pas été effectuées dans la musculature cervicale ont aussi été rapportés (pour d'autres informations, voir la section «Précautions à prendre selon les indications», «Dystonie cervicale»).
BOTOX ne doit être administré qu'avec une extrême prudence et sous contrôle strict chez les patients atteints d'un trouble de la conduction neuromusculaire subclinique ou clinique avéré, comme p.ex. en cas de myasthénie grave ou de syndrome de Eaton-Lambert, chez les patients souffrant de maladies motrices neuropathiques périphériques (p.ex. sclérose latérale amyotrophique ou neuropathie motrice) et chez les patients avec des maladies neurologiques de base. Ces patients peuvent présenter une sensibilité accrue aux principes actifs tels que BOTOX, également aux doses thérapeutiques, ce qui peut entraîner une faiblesse musculaire prononcée et un risque accru d'effets systémiques cliniquement significatifs, y compris de dysphagie sévère et de trouble respiratoire. La préparation de toxine botulique ne doit, chez ces patients, être utilisée que sous la surveillance par un spécialiste et seulement lorsque les bénéfices du traitement dépassent les risques associés. Les patients avec des antécédents de dysphagie et d'aspiration doivent être traités avec la plus grande prudence.
Il convient d'informer les patients et le personnel soignant de la nécessité de contacter immédiatement le médecin en cas d'apparition de troubles de la déglutition, de l'élocution ou de la respiration.
Il faut conseiller aux patients sortant d'une période de sédentarité de reprendre progressivement et prudemment l'activité physique.
Avant d'utiliser BOTOX, le médecin doit se familiariser avec l'anatomie du patient et avec les changements anatomiques dus à des interventions chirurgicales. Il faut éviter les injections dans des structures anatomiques fragiles.
Après l'utilisation de BOTOX à proximité du thorax, un pneumothorax en relation avec l'injection a été rapporté. Il convient d'être prudent en cas d'injection à proximité des poumons (en particulier l'apex) ou dans d'autres structures fragiles.
Des effets indésirables sévères, comprenant des effets avec issue fatale, ont été rapportés chez des patients ayant reçu BOTOX directement dans les glandes salivaires, la sphère orolinguale, l'œsophage ou l'estomac (indications non autorisées (off-label)). Certains patients souffraient déjà d'une dysphagie ou étaient dans un état de faiblesse avancé.
De rares réactions d'hypersensibilité graves et/ou immédiates y compris choc anaphylactique, maladie sérique, urticaire, œdème des parties molles et dyspnée ont été rapportées. Quelques-unes de ces réactions sont survenues soit après utilisation de BOTOX seul, soit après utilisation de BOTOX combiné à d'autres préparations dont l'utilisation s'accompagne de réactions similaires. Si une telle réaction survient, il faut renoncer à une autre injection de BOTOX et instaurer immédiatement un traitement médical adéquat, p.ex. adrénaline. Dans un cas, une anaphylaxie avec issue fatale a été rapportée chez un patient ayant reçu une injection de BOTOX reconstituée par inadvertance dans 5 ml d'une solution de lidocaïne à 1%.
Comme pour toute injection, une lésion inhérente à l'administration du médicament peut survenir. Une injection peut conduire à une infection locale, des douleurs, des inflammations, une paresthésie, une hypoesthésie, une sensibilité, une enflure, un érythème et/ou un saignement/une ecchymose. Des douleurs et/ou l'anxiété en rapport avec l'aiguille peuvent induire des réactions vasovagales, p.ex. syncope, hypotension, etc.
La prudence s'impose lorsque BOTOX est utilisé en présence d'une inflammation au(x) site(s) d'injection proposé(s) ou lorsque le muscle cible présente une faiblesse excessive ou une atrophie. La prudence s'impose également lorsque BOTOX est utilisé pour le traitement de patients atteints de maladies motrices neuropathies périphériques (p.ex. sclérose latérale amyotrophique ou neuropathie motrice).
Des effets indésirables touchant le système cardiovasculaire comme l'arythmie et l'infarctus du myocarde, parfois avec issue fatale, ont été rapportés après l'utilisation de BOTOX. Certains de ces patients présentaient des facteurs de risque comme des maladies cardiovasculaires.
Des crises d'épilepsie nouvelles ou répétées ont été rapportées, particulièrement chez des patients avec une prédisposition pour de telles manifestations. La relation précise entre ces cas et l'injection de toxine botulique n'est pas prouvée. Les rapports d'effets indésirables chez les enfants concernent surtout des patients avec parésie cérébrale infantile traités contre la spasticité.
La formation d'anticorps neutralisants dirigés contre la toxine botulique de type A peut réduire l'efficacité d'un traitement par BOTOX en bloquant l'activité biologique de la toxine. Des résultats de quelques études laissent supposer que l'administration d'injections de BOTOX à intervalles plus fréquents ou à des doses plus élevées peut conduire à une augmentation de l'incidence de la formation d'anticorps. Le cas échéant, la formation d'anticorps potentielle peut être réduite par l'administration de la dose efficace la plus faible et par le maintien d'un intervalle le plus long possible tout en restant cliniquement approprié entre les injections.
Lors de l'utilisation répétée de BOTOX (comme avec toutes les toxines botuliques), des variations cliniques peuvent survenir en raison des différences entre les procédures de reconstitution des flacons, des intervalles d'injection, des muscles traités et de légères différences d'activité de la toxine qui sont dues aux essais biologiques utilisés.
Après un contact de BOTOX avec la peau, la zone de peau concernée doit d'abord être nettoyée avec une solution diluée d'hypochlorite, puis rincée abondamment à l'eau courante. Dans le cas d'une piqûre accidentelle, la zone de peau concernée doit également être nettoyée immédiatement et le patient doit être surveillé comme recommandé dans la rubrique «Surdosage». En cas de contact avec les yeux, l'œil concerné doit être rincé abondamment à l'eau courante ou avec une solution appropriée.
Si une contamination accidentelle est suspectée (p.ex. contact avec la peau ou piqûre accidentelle), la zone de peau doit immédiatement être nettoyée. Le patient doit être surveillé médicalement comme recommandé dans le paragraphe «Surdosage».
Utilisation chez les enfants et les adolescents
L'innocuité et l'efficacité de BOTOX ne sont pas prouvées pour des indications autres que celles mentionnées pour les enfants et les adolescents dans la rubrique «Indications/Possibilités d'emploi». Des cas de diffusion éventuelle de la toxine dans des zones éloignées du site d'injection ont été très rarement rapportés après la commercialisation du produit chez des enfants et des adolescents présentant une maladie supplémentaire. Ces cas concernaient surtout des patients présentant des parésies cérébrales infantiles. En général, la posologie utilisée dans ces cas était supérieure à celle recommandée (voir «Effets indésirables»).
Des décès ont été rarement rapportés chez des enfants souffrant de parésie cérébrale sévère après un traitement par la toxine botulique, parfois en relation avec une pneumonie d'aspiration et parfois après une utilisation non autorisée (off-label) (p.ex. au niveau de la nuque). Des précautions extrêmes sont indispensables lors du traitement des enfants et des adolescents en état de faiblesse neurologique sévère, de dysphagie ou avec des antécédents de pneumonie d'aspiration ou de maladie pulmonaire. Le traitement chez les patients en mauvaise santé ne doit avoir lieu que si le médecin estime que les bénéfices éventuels dépassent les risques en fonction de la situation individuelle.
Ce médicament contient moins de 1 mmol de sodium (23 mg) par unité de dosage, c'est-à-dire qu'il est pratiquement «sans sodium».
Précautions à prendre selon les indications
AFFECTIONS NEUROLOGIQUES:
Blépharospasme
Une diminution des clignements après l'injection de toxine botulique dans le muscle orbiculaire de l'œil peut conduire à une sollicitation intense de la cornée, des défauts épithéliaux durables et des ulcérations de la cornée, surtout chez les patients avec des troubles du nerf cérébral VII. Il convient d'effectuer des tests minutieux de sensibilité cornéenne sur les yeux précédemment opérés, d'éviter toute injection dans la zone médiale de la paupière inférieure pour prévenir la formation d'ectropion et de traiter activement toute lésion épithéliale. Ces traitements peuvent nécessiter l'emploi d'un collyre protecteur, d'une pommade, de lentilles souples thérapeutiques ou l'occlusion de l'œil par un cache-œil ou un autre moyen.
Des ecchymoses sont possibles dans les tissus mous de la paupière. Il est possible de minimiser ce phénomène en appliquant une pression légère au point d'injection après l'injection.
En raison de l'activité anticholinergique de la toxine botulique, la prudence s'impose chez les patients présentant un glaucome à angle fermé, y compris les patients atteints d'un angle de la chambre antérieure anatomiquement étroit.
Strabisme
BOTOX est inefficace dans le traitement du strabisme paralytique chronique, sauf pour réduire une contracture du muscle antagoniste en relation avec une correction chirurgicale. Lors de l'utilisation de BOTOX pour traiter le strabisme, un saignement rétrobulbaire ou une perforation du bulbe peuvent se produire.
Dystonie cervicale
En cas de dystonie cervicale, des injections de BOTOX peuvent causer une dysphagie très légère à sévère. Consécutivement à la dysphagie, une aspiration et une dyspnée sont possibles et peuvent entraîner dans des cas très rares la nécessité d'une alimentation artificielle. On a signalé de rares cas de dysphagies associées à une pneumonie d'aspiration et la mort. La dysphagie peut durer pendant 2 à 3 semaines après l'injection mais des cas de dysphagie durant jusqu'à 5 mois ont été rapportés.
La fréquence des dysphagies est dose-dépendante et il est possible de la réduire en maintenant la dose injectée dans le muscle sterno-cléido-mastoïdien inférieure à 100 unités. Les patients présentant une faible masse musculaire au niveau de la nuque ou ayant reçu une injection bilatérale dans le muscle sterno-cléido-mastoïdien présentent un risque de dysphagie plus élevé. La dysphagie est imputable à la diffusion de la toxine dans la musculature œsophagienne. Les patients souffrant de dystonie cervicale doivent être informés du risque de dysphagie en tant qu'effet indésirable.
Des injections dans l'élévateur de l'omoplate peuvent s'accompagner d'une augmentation du risque d'infection des voies respiratoires supérieures et de dysphagie.
Une dysphagie peut contribuer à une diminution de la prise de nourriture et d'eau, ce qui conduit à une perte de poids et à une déshydratation. Les patients avec une dysphagie subclinique peuvent présenter un risque accru de dysphagie sévère après une injection de BOTOX.
Spasticités focales
BOTOX est un traitement des spasticités focales étudié seulement en association avec des traitements standards qu'il ne vise pas à remplacer.
Il est peu probable qu'il puisse améliorer la mobilité d'une articulation touchée par une contracture fixée.
BOTOX ne doit pas être utilisé pour le traitement de la spasticité focale de la cheville et du pied chez les adultes ayant subi un AVC si l'on ne s'attend pas à une amélioration de la fonction (p. ex., amélioration de la marche) ou à un soulagement des symptômes (p. ex., soulagement de la douleur) ou à une facilitation des soins en raison de la réduction du tonus musculaire.
Il convient d'être particulièrement vigilant lors du traitement des adultes souffrant de spasticité suite à un AVC qui peuvent présenter un risque accru de chute.
BOTOX doit être utilisé avec prudence en cas de spasticité focale de la cheville et du pied chez les patients âgés ayant été victimes d'un AVC et présentant une comorbidité marquée. Le traitement ne doit être initié que si l'on considère que les bénéfices du traitement l'emportent sur les risques éventuels.
BOTOX ne doit être utilisé pour le traitement de la spasticité des membres inférieurs après un AVC qu'après évaluation par des médecins expérimentés dans la gestion de la rééducation des patients victimes d'un AVC.
Il existe des rapports post-commercialisation signalant des cas de décès suite à un traitement par la toxine botulique (dans certains cas dus à une pneumonie d'aspiration) et une diffusion possible de la toxine dans des zones éloignées du site d'application chez des enfants souffrant de maladies supplémentaires, en particulier d'infirmité motrice cérébrale. Voir les mises en garde concernant «l'utilisation chez les enfants et les adolescents» dans la rubrique «Mises en garde et précautions». Un rapport causal avec BOTOX n'a pas été démontré dans les cas mentionnés.
Céphalées en cas de migraine chronique:
La sécurité et l'efficacité n'ont pas été établies à ce jour dans la prophylaxie des céphalées chez les patients atteints de migraine épisodique (céphalées pendant < 15 jours par mois) ou de céphalées de tension chroniques.
DYSFONCTIONS VÉSICALES:
Injections dans le muscle détrusor
Les précautions médicales d'usage doivent être mises en œuvre lors de la cystoscopie.
Chez les patients qui n'ont pas recours au sondage vésical, le volume urinaire résiduel post-mictionnel doit être évalué dans les 2 semaines suivant le traitement, puis de façon périodique selon avis médical durant une période allant jusqu'à 12 semaines. Les patients doivent être informés de la nécessité de contacter leur médecin en cas de difficultés lors de la miction, car dans ce cas, un sondage vésical est éventuellement nécessaire.
Vessie hyperactive
BOTOX doit être utilisé avec précautions chez les patients atteints d'obstructions dans la région du col vésical (p.ex. obstructions des voies urinaires chez les patients atteints d'une hyperplasie de la prostate).
Incontinence urinaire causée par l'hyperactivité neurogène du détrusor
Une hyper-refléxie autonome associée à la procédure peut survenir. Dans ce cas, un traitement médical immédiat peut être nécessaire.
AFFECTIONS DE LA PEAU ET APPARENTÉES:
Hyperhidrose axillaire primaire
Il convient de rechercher les causes éventuelles d'une hyperhidrose secondaire (p.ex. hyperthyroïdie, phéochromocytome), afin d'éviter le traitement symptomatique d'une hyperhidrose, sans diagnostic et/ou traitement de la pathologie sous-jacente.
Excipients pharmaceutiques revêtant un intérêt particulier
Ce médicament contient moins de 1 mmol de sodium (23 mg) par flacon, c'est-à-dire qu'il est essentiellement «sans sodium».
InteractionsThéoriquement l'effet de la toxine botulique de type A peut-être potentialisé par la co-administration de BOTOX avec des antibiotiques aminoglycosides, la spectinomycine ou d'autres agents pouvant interférer avec la transmission neuromusculaire (p.ex. relaxants musculaires).
L'effet de l'administration simultanée ou à plusieurs mois d'intervalle de différents sérotypes de toxine botulique est inconnu. Une faiblesse neuromusculaire marquée peut être exacerbée par l'administration d'une autre toxine botulique avant la résolution complète des effets d'une toxine botulique préalablement administrée.
Les myorelaxants doivent être utilisés avec prudence.
Aucune étude d'interaction n'a été effectuée. Aucune interaction d'importance clinique n'a été rapportée.
Enfants et adolescents
Aucune étude pour l'enregistrement des interactions n'a été réalisée chez l'enfant et l'adolescent.
Il convient toutefois de noter que les interactions mentionnées ci-dessus peuvent également se produire dans la population pédiatrique.
Grossesse, allaitementGrossesse
Il n'existe pas de données suffisantes concernant l'utilisation de la toxine botulique de type A pendant la grossesse. Les études réalisées sur les animaux ont mis en évidence un effet de toxicité reproductive (voir section «Données précliniques»).
Le risque potentiel pour l'homme est inconnu. BOTOX ne doit pas être utilisé pendant la grossesse ou chez des femmes en âge de procréer qui n'utilisent pas de méthode de contraception, sauf en cas de nécessité absolue. Si la patiente tombe enceinte pendant le traitement, elle doit être informée des risques potentiels de fausse couche et de malformation congénitale.
Allaitement
On ne sait pas si BOTOX passe dans le lait maternel. L'utilisation de BOTOX pendant l'allaitement ne peut pas être recommandée.
Fertilité
Il n'existe pas de données concernant l'utilisation de la toxine botulique de type A. Les études expérimentales réalisées sur des animaux avec BOTOX ont mis en évidence des effets sur la fertilité masculine et féminine (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude concernant les effets sur l'aptitude à la conduite et l'utilisation des machines n'a été effectuée. BOTOX peut cependant provoquer une asthénie, une faiblesse musculaire, des vertiges et des troubles visuels pouvant influencer l'aptitude à participer activement au trafic routier et à utiliser des machines.
Effets indésirablesEn général
Des effets indésirables, qui, de l'avis des médecins investigateurs étaient en rapport avec BOTOX, ont été rapportés dans des études cliniques contrôlées: chez 35% des patients avec blépharospasme, chez 28% des patients avec dystonie cervicale, chez 8% des patients avec spasticité infantile, chez 11% des patients avec hyperhidrose axillaire primaire, chez 16% des patients adultes avec spasticité des membres supérieurs et chez 11% des patients adultes atteints de spasticité des membres inférieurs. Dans des études cliniques portant sur la vessie hyperactive, la fréquence était de 26% lors du premier traitement et de 22% lors du second traitement.
Dans des études cliniques relatives à l'incontinence urinaire causée par l'hyperactivité neurogène du détrusor, la fréquence était de 32% après le premier traitement, puis de 18% après le second traitement.
Dans les études cliniques sur l'hyperactivité neurogène du détrusor chez les adultes, la fréquence était de 32 % lors du premier traitement et a baissé à 18 % lors du deuxième traitement. Dans le cas de l'hyperactivité neurogène du détrusor pédiatrique, l'incidence s'élevait à 14,2 % (16/113) lors du premier traitement, 16,7 % (15/90) lors du deuxième traitement, 18,2 % (10/55) lors du troisième traitement et 45,5 % (5/11) lors du quatrième traitement.
Dans les études cliniques conduites dans la migraine chronique, la fréquence était de 26 % après le premier traitement et diminuait à 11 % après le deuxième traitement.
En général, les effets secondaires s'observent dans les premiers jours suivant l'injection et peuvent persister pendant plusieurs mois ou, dans des cas rares, plus longtemps, bien qu'en général ils soient transitoires.
L'action pharmacologique attendue de la toxine botulique est une faiblesse musculaire locale. Cependant la faiblesse des muscles adjacents et/ou des muscles à distance du site d'injections a été rapportée.
Comme pour toute injection, on peut observer une douleur locale, une inflammation, une paresthésie, une hypoesthésie, une sensibilité à la pression, un gonflement/œdème, un érythème, une infection locale, des saignements et/ou épanchements sanguins sont survenus au site d'injection. Des douleurs dues à l'aiguille d'injection et/ou des sentiments d'angoisse ont entraîné des réactions vasovagales, y compris une hypotension symptomatique transitoire et une syncope. De la fièvre et des symptômes grippaux ont aussi été rapportés après des injections de toxine botulique.
Effets secondaires – fréquence par indication
Les paragraphes suivants contiennent, en fonction des domaines d'indications, des données sur la fréquence des effets secondaires documentés dans des études cliniques.
Les fréquences sont définies comme suit: très fréquents (≥1/10); fréquents (≥1/100 à <1/10); occasionnels ( ≥1/1000 à <1/100); rares (≥1/10'000 à <1/1000); très rares (<1/10'000), fréquence inconnue (ne peut être estimée sur la base des données disponibles).
AFFECTIONS NEUROLOGIQUES:
Indication Blépharospasme/hémispasme facial
Affections du système nerveux
Occasionnels: vertiges, parésie faciale, paralysie faciale.
Affections oculaires
Très fréquents: ptose de la paupière supérieure (11%).
Fréquents: kératite ponctuée, lagophtalmie, sécheresse oculaire, irritation oculaire, photophobie, larmoiement augmenté.
Occasionnels: kératite, ectropion, diplopie, entropion, troubles visuels, vision trouble.
Rares: œdème palpébral.
Très rares: kératite ulcéreuse, altération de l'épithélium cornéen, perforation de la cornée.
Affections de la peau et du tissu sous-cutané
Fréquents: ecchymose.
Occasionnels: exanthème, dermatite.
Troubles généraux et anomalies au site d'administration
Fréquents: œdème facial, irritations.
Occasionnels: asthénie.
Indication Strabisme
Affections oculaires
Très fréquents: troubles du mouvement des yeux (16,9%), ptose de la paupière supérieure (15,7%).
Occasionnels: hémorragies oculaires rétrobulbaires, pénétration des yeux, pupille d'Adie-Holmes.
Rares: hémorragie du corps vitré.
Indication Dystonie cervicale
Infections et infestations
Fréquents: rhinite, infection des voies respiratoires supérieures.
Affections du système nerveux
Fréquents: vertiges, céphalées, hypoesthésie, hypertonie musculaire, somnolence.
Affections oculaires
Occasionnels: diplopie, ptose de la paupière supérieure.
Affections respiratoires, thoraciques et médiastinales
Occasionnels: dyspnée, dysphonie.
Affections gastro-intestinales
Très fréquents: dysphagie (jusqu'à 18,6% des patients, avec une dose moyenne de 240,5 unités) (voir également «Informations supplémentaires» plus bas).
Fréquents: bouche sèche, nausées.
Affections musculo-squelettiques et du tissu conjonctif
Très fréquents: faiblesse musculaire (12,8%).
Fréquents: raideur de la musculature squelettique, courbatures.
Troubles généraux et anomalies au site d'administration
Très fréquents: douleur (16,3%).
Fréquents: asthénie, symptômes grippaux, malaise.
Occasionnels: fièvre.
Indication Spasticité focale des extrémités supérieures - Adultes
Affections psychiatriques
Occasionnels: dépression, insomnie.
Affections du système nerveux
Fréquents: hypertonie musculaire.
Occasionnels: paresthésie, céphalées, hypoesthésie, amnésie, troubles de la coordination.
Affections de l'oreille et du labyrinthe
Occasionnels: vertiges.
Affections vasculaires
Occasionnels: hypotension orthostatique.
Affections gastro-intestinales
Occasionnels: nausées, paresthésie orale.
Affections de la peau et du tissu sous-cutané
Fréquents: ecchymoses, purpura.
Occasionnels: dermatite, prurit, exanthème.
Affections musculo-squelettiques et du tissu conjonctif
Fréquents: douleur dans les extrémités, faiblesse musculaire.
Occasionnels: arthralgie, bursite.
Troubles généraux et anomalies au site d'administration
Fréquents: douleurs au site d'injection, fièvre, symptômes grippaux, saignements ou irritations au site d'injection.
Occasionnels: douleurs, sensation de malaise, asthénie, hypersensibilité au site d'administration, œdème périphérique.
Certains des effets indésirables rapportés occasionnellement peuvent être dus à la maladie.
Indication Spasticité focale des extrémités supérieures - Adolescents et enfants à partir de 2 ans
Infections et affections parasitaires
Fréquentes: infections des voies respiratoires supérieures.
Affections du tractus gastro-intestinal
Fréquentes: nausées.
Troubles des muscles squelettiques, du tissu conjonctif et des os
Fréquente: faiblesse musculaire.
Troubles généraux et anomalies au site d'administration
Fréquentes: douleurs au site d'injection.
Indication Spasticité focale des membres inférieurs - Adultes
Troubles des muscles squelettiques, du tissu conjonctif et des os
Fréquente: arthralgie.
Troubles généraux et anomalies au site d'administration
Fréquent: œdème périphérique.
Aucune différence sur le plan du profil de la sécurité n'a été observée lors de l'administration de doses répétées.
Indication Spasticité focale des membres inférieurs - Adolescents et enfants à partir de 2 ans
Troubles généraux et anomalies au site d'administration
Fréquentes: douleurs au site d'injection.
Céphalées en cas de migraine chronique
Affections du système nerveux
Fréquents: céphalées, migraine, parésie faciale.
Affections oculaires
Fréquents: ptose de la paupière supérieure.
Affections gastro-intestinales
Occasionnels: dysphagie.
Affections de la peau et du tissu sous-cutané
Fréquents: prurit, éruption cutanée.
Occasionnels: douleurs cutanées.
Affections musculosquelettiques et du tissus conjonctif
Fréquents: douleur cervicale, myalgie, douleurs de l'appareil locomoteur, raideur de l'appareil locomoteur, spasmes musculaires, contractures musculaires, faiblesse musculaire
Occasionnels: douleurs dans la mâchoire.
Fréquence inconnue: signe de Méphisto (élévation latérale des sourcils).
Troubles généraux et anomalies au site d'administration
Fréquents: douleurs au site d'injection.
Une migraine, y compris une aggravation de la migraine, a été rapportée chez 3,8 % des patients sous BOTOX et 2,6 % des patients sous placebo, survenant généralement au cours du premier mois suivant le traitement.
Ces réactions ne se sont pas reproduites systématiquement lors des cycles de traitement suivants et l'incidence globale a diminué lors de traitements répétés.
Le taux d'arrêt du traitement en raison d'événements indésirables, dans ces études de phase 3, était de 3,8 % sous BOTOX contre 1,2% sous placebo.
AFFECTIONS VÉSICALES:
Indication Hyperactivité vésicale - adultes
Infections et infestations
Très fréquents: infections des voies urinaires (25,5%).
Fréquents: bactériurie.
Affections du rein et des voies urinaires
Très fréquents: dysurie (10,9%).
Fréquents: rétention urinaire, pollakiurie.
Investigations
Fréquents: volume d'urine restante*.
* Volume d'urine restante accru après vidange de la vessie qui ne nécessite pas de cathétérisme.
Les effets secondaires fréquents dus à la procédure étaient la dysurie et l'hématurie.
Un cathétérisme intermittent propre a été instauré chez 6,5% des patients après le traitement par 100 unités de BOTOX contre 0,4% dans le groupe placebo.
Parmi les 1'242 patients participant à des études cliniques contrôlées contre placebo, 41,4% (n = 514) avaient ≥65 ans et 14,7% (n = 182) ≥75 ans. Dans l'ensemble, aucune différence sur le plan du profil de la sécurité n'a été constatée après le traitement par BOTOX entre les patients de 65 ans et plus et les patients de moins de 65 ans, à l'exception du fait que les infections des voies urinaires se sont manifestées plus fréquemment chez les patients plus âgés, tant dans le groupe placebo que dans le groupe BOTOX, que chez les patients plus jeunes.
Dans l'ensemble, aucune modification du profil de la sécurité n'a été observée lors de l'administration répétée.
Hyperactivité vésicale chez les enfants et les adolescents
Infections et infestations
Fréquents: infection des voies urinaires.
Affections des reins et des voies urinaires
Fréquents: dysurie*, douleur urétrale*.
*Effets indésirables liés à la procédure
Affections gastro-intestinales
Fréquents: douleurs abdominales, douleurs au niveau du bas-ventre.
Dans une étude en double aveugle, randomisée et multi-centrique à groupes parallèles portant sur 55 patients âgés de 12 à 17 ans, les effets indésirables étaient comparables au profil de tolérance connu chez les patients adultes présentant une hyperactivité vésicale, mais des douleurs urétrales et abdominales ont également été identifiées lors de cette petite étude de l'hyperactivité vésicale pédiatrique.
Indication Incontinence urinaire chez les adultes à la suite d'une hyperactivité neurogène du détrusor
Effets indésirables à l'issue des études cliniques de phase 2 et des études cliniques pivots de phase 3
Infections et infestations
Très fréquents: infections des voies urinaires (49%).
Affections psychiatriques
Fréquents: insomnie.
Affections gastro-intestinales
Fréquents: constipation.
Affections musculo-squelettiques et du tissu conjonctif
Fréquents: faiblesse musculaire, spasmes musculaires.
Affections du rein et des voies urinaires
Très fréquents: rétention urinaire (17%).
Fréquents: hématurie*, dysurie*, diverticules vésicaux.
Troubles généraux et anomalies au site d'administration
Fréquents: fatigue, troubles de la marche.
Lésions, intoxications et complications liées aux procédures
Fréquents: hyper-réflexie* autonome, chute.
* Effets indésirables liés à la procédure
Dans les études cliniques, des infections des voies urinaires ont été rapportées chez 49,2% des patients ayant été traités par 200 unités de BOTOX et chez 35,7% des patients ayant été traités par un placebo (53,0% des patients avec sclérose en plaques (SEP) ayant reçu 200 unités contre 29,3% dans le groupe placebo; 45,4% des patients atteints de lésions médullaires ayant été traités par 200 unités contre 41,7% de ceux ayant été traités par placebo). Des rétentions urinaires ont été rapportées chez 17,2% des patients ayant été traités par 200 unités de BOTOX, ainsi que chez 2,9% de ceux traités par placebo (28,8% des patients avec sclérose en plaques ayant reçu 200 unités contre 4,5% dans le groupe placebo; 5,4% des patients atteints de lésions médullaires ayant été traités par 200 unités contre 1,4% de ceux ayant été traités par placebo).
Lors d'administrations répétées, aucune modification de la nature des effets indésirables n'a été rapportée.
Chez les patients avec SEP ayant participé aux études pivots, aucune modification du taux annuel de poussée de SEP (c.-à-d. le nombre de poussées de SEP par année-patient) n'a été observée (BOTOX = 0,23; placebo = 0,20).
Parmi les patients qui n'avaient pas recours à un sondage vésical au début de l'étude, avant le traitement, un sondage a été instauré dans 38,9% des cas après le traitement par BOTOX 200 unités contre 17,3% des cas chez les patients recevant le placebo.
Effets indésirables à l'issue de l'étude réalisée après l'autorisation de mise sur le marché avec 100 unités de BOTOX sur des patients SEP non cathétérisés en début d'étude
Infections et infestations
Très fréquents: infections des voies urinaires (39,4%), bactériurie (18,2%).
Affections du rein et des voies urinaires
Très fréquents: rétention urinaire (16,7%).
Fréquents: hématurie**, dysurie**.
Investigations
Très fréquents: volume d'urine restante** (16,7%)
* Effets indésirables liés à la procédure
** Volume d'urine restante accru qui ne nécessite pas de cathétérisme
Un sondage a été instauré chez 15,2% des patients après le traitement avec 100 unités de BOTOX par rapport à 2,6% des patients recevant le placebo.
Indication Hyperactivité neurogène du détrusor chez les patients pédiatriques à partir de 5 ans
Effets indésirables (toutes doses) issus de deux études de phase 3 en pédiatrie, y compris cycle de traitement 1 de l'étude 191622-120 (N = 113) et cycles de traitement répétés 2 (N = 90), 3 (N = 55) et 4 (N = 11) de l'étude 191622-121:
Infections et infestations
Très fréquents: Infection des voies urinaires (cycle 1 = 29,2 %, cycle 2 = 34,4 %, cycle 3 = 21,8 %, cycle 4 = 18,2 %), bactériurie (cycle 1 = 16,8 %, cycle 2 = 13,3 %, cycle 3 = 12,7 %, cycle 4 = 18,2 %).
Affections du rein et des voies urinaires
Très fréquents: Hématurie ou présence de sang dans les urines (cycle 1 = 4,4 %, cycle 2 = 10,0 %, cycle 3 = 16,4 %, cycle 4 = 54,5%).
Fréquents: Leucocyturie.
Affections gastro-intestinales
Fréquents: Constipation, douleurs abdominales*
Troubles généraux et anomalies au site d'administration
Fréquents: Douleurs supra pubiennes*.
* Effets indésirables provoqués par la procédure
AFFECTIONS DE LA PEAU ET DU TISSU SOUS-CUTANÉ:
Indication Hyperhidrose axillaire primaire
Affections du système nerveux
Fréquents: douleurs, céphalées, paresthésie.
Affections vasculaires
Fréquents: bouffées de chaleur.
Affections gastro-intestinales
Occasionnels: nausées.
Affections de la peau et du tissu sous-cutané
Fréquents: hyperhidrose (sudation non-axillaire), odeur anormale de la peau, prurit, nodule sous-cutané, alopécie.
Affections musculo-squelettiques et du tissu conjonctif
Fréquents: douleurs dans les extrémités.
Occasionnels: faiblesse des bras, myalgie, arthropathie.
Troubles généraux et anomalies au site d'administration
Très fréquents: douleurs au site d'injection (11,5%).
Fréquents: douleurs, œdème/saignement/hypersensibilité/irritation au site d'administration, asthénie, réactions au site d'administration.
Lors du traitement de l'hyperhidrose axillaire primaire, une augmentation de la production de sueur en dehors des aisselles a été rapportée chez 4,5% des patients dans le mois suivant l'injection. Ce faisant, un schéma relatif aux sites anatomiques n'a pas été défini. Chez environ 30% des patients, une régression a pu être constatée dans les 4 mois.
Occasionnellement (0,7%) une légère faiblesse transitoire des bras ne nécessitant cependant pas de traitement et qui s'est améliorée sans séquelles a été rapportée. Cet effet secondaire pourrait être dû au traitement, à la technique d'injection ou aux deux. Lors de la faiblesse musculaire rapportée occasionnellement, un examen neurologique peut être envisagé. En outre, un contrôle de la technique d'injection est indiqué avant les injections suivantes afin d'assurer le placement intradermique des injections.
Effets indésirables après commercialisation
La liste suivante indique des effets médicamenteux indésirables ou d'autres effets indésirables médicalement pertinents rapportés indépendamment de l'indication depuis la commercialisation du médicament, en plus des effets indésirables mentionnés dans la rubrique «Mises en garde et précautions» et la rubrique «Effets indésirables».
Affections du système immunitaire
Anaphylaxie, angio-œdème, maladie sérique et urticaire.
Troubles du métabolisme et de la nutrition
Anorexie.
Affections du système nerveux
Plexopathie brachiale, dysphonie, dysarthrie, parésie faciale, hypoesthésie, faiblesse musculaire, myasthénie grave, neuropathie périphérique, paresthésie, radiculopathie, convulsions, syncope et paralysie faciale.
Affections oculaires
Glaucome à angle étroit (lors du traitement du blépharospasme), strabisme, vue trouble, troubles de la vue, sécheresse oculaire et œdème palpébral.
Affections de l'oreille et du labyrinthe
Hypoacousie, acouphène et vertiges.
Affections cardiaques
Arythmie, infarctus du myocarde.
Affections respiratoires, thoraciques et médiastinales
Pneumonie d'aspiration (avec parfois une issue fatale), dyspnée, dépression respiratoire et insuffisance respiratoire.
Affections gastro-intestinales
Douleurs abdominales, diarrhée, constipation, sécheresse buccale, dysphagie, nausées et vomissements.
Affections de la peau et du tissu sous-cutané
Alopécie, dermatite de type psoriasis, érythème multiforme, hyperhidrose, madarosis, prurit et éruption cutanée.
Troubles musculo-squelettiques et du tissu conjonctif
Atrophie musculaire, myalgie et contractions musculaires localisées/contractions musculaires involontaires.
Troubles généraux et anomalies au site d'administration
Atrophie par dénervation, malaise et fièvre.
Dans le traitement du strabisme, on a enregistré un cas de saignement du corps vitré, qui s'est résorbé par la suite. On connaît certains cas d'hémorragies rétrobulbaires, sans perte de vision cependant. Cinq yeux ont présenté des modifications pupillaires, avec une lésion du ganglion ciliaire (pupille d'Adie).
Une patiente a développé une plexopathie brachiale deux jours après avoir reçu par injection 120 unités de BOTOX pour le traitement d'une dystonie cervicale, la guérison intervenant cinq mois après.
Chez des patients atteints de spasticité des extrémités supérieures après un AVC, on a réalisé des mesures de spirométrie pulmonaire indiquant une diminution de 15% de la fonction pulmonaire. L'incidence de cet effet indésirable a été plus importante parmi le groupe de patients ayant reçu BOTOX que dans le groupe ayant reçu un placebo, même si les différences n'étaient ni cliniquement, ni statistiquement, significatives.
Dans le traitement de l'hyperhidrose axillaire primaire, on a signalé une augmentation perçue de la sudation non-axillaire chez 4,5% des patients dans le mois suivant l'injection, sans lien systématique avec les sites anatomiques touchés. On a constaté une résolution de ce trouble chez 30% environ des patients au bout de quatre mois.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageLe surdosage de BOTOX est un terme relatif et dépend de la dose, du site d'injection et des propriétés tissulaires sous-jacentes. Aucun cas de toxicité systémique résultant d'une injection accidentelle de solution de BOTOX n'a été observé. Des surdosages peuvent causer des paralysies neuromusculaires locales ou éloignées, générales et profondes. Aucun cas d'ingestion de BOTOX n'a été rapporté.
Les signes et symptômes d'un surdosage ne se manifestent pas immédiatement après une injection. En cas d'injection accidentelle ou d'ingestion ou de surdosage suspecté, le patient doit être soumis à une surveillance médicale durant plusieurs semaines à la recherche des signes et symptômes croissants d'une faiblesse musculaire locale ou éloignée du site d'injection, comme la ptose, la diplopie, la dysphagie, la dysarthrie, la faiblesse généralisée ou l'insuffisance respiratoire. Chez ces patients, on considérera des investigations complémentaires et initiera immédiatement un traitement médical approprié, éventuellement avec hospitalisation du patient.
Si la musculature de l'oropharynx et de l'œsophage est touchée, une pneumonie d'aspiration peut se manifester. Si une paralysie ou un affaiblissement marqué des muscles respiratoires survient, une intubation et une assistance respiratoire seront nécessaires jusqu'au rétablissement et une trachéostomie ainsi qu'une respiration artificielle durant une longue période peuvent s'avérer nécessaires en plus des mesures de soutien générales.
Propriétés/EffetsCode ATC
M03AX01
Mécanisme d'action
La toxine botulique de type A bloque la libération d'acétylcholine périphérique au niveau des terminaisons nerveuses cholinergiques présynaptiques par clivage de la SNAP-25. Cette dernière est une protéine synaptique de 25 kDa, nécessaire à l'ancrage et à la libération de l'acétylcholine par les vésicules situées au sein des terminaisons nerveuses.
Pharmacodynamique
Après l'injection, il se produit une liaison initiale rapide et de haute affinité entre la toxine et des récepteurs cellulaires de surface spécifiques. Celle-ci est suivie du transfert de la toxine à travers la membrane plasmique par endocytose médiée par les récepteurs. Enfin, la toxine est libérée dans le cytoplasme. Ce processus est accompagné d'une inhibition progressive de la libération d'acétylcholine. Les signes cliniques se manifestent dans un délai de 2 à 3 jours, avec un effet maximal constaté 5 à 6 semaines après l'injection. Au niveau des neurones sensoriels, BOTOX inhibe la libération des neurotransmetteurs sensoriels (p.ex. substance P, CGRP) et réduit l'expression des récepteurs de surface cellulaires (p.ex. TRPV1). De plus, BOTOX empêche et inverse la sensibilisation des neurones sensoriels nociceptifs, ce qui peut donc éventuellement aussi inhiber la sensibilisation centrale.
Après une injection intramusculaire, la transmission de l'influx nerveux se rétablit normalement en l'espace de 12 semaines, lorsque les terminaisons nerveuses repoussent et se reconnectent avec les plaques motrices.
Après une injection intradermique visant les glandes sudoripares écrins, l'effet dure environ 4 à 7 mois chez des patients traités par 50 unités par aisselle.
En utilisant BOTOX pour traiter le strabisme, on suppose que ce produit agit sur des paires de muscles dans lesquelles le muscle traité est soumis à un étirement atrophique et le muscle antagoniste correspondant est raccourci.
Après injection dans la musculature de la nuque, BOTOX améliore les signes objectifs et les symptômes subjectifs de la dystonie cervicale. Cette amélioration se manifeste par une réduction de la torsion latérale de la tête, un moindre soulèvement des épaules, un volume et une force plus faibles des muscles hypertrophiés, ainsi qu'une atténuation de la douleur.
Chez l'adulte atteint de spasticité focale des extrémités supérieures après un AVC, BOTOX améliore de façon importante la mobilité (coude et poignet).
Chez les enfants présentant une déformation dynamique du pied en équin sans rétraction et sans atrophie importante, l'injection de BOTOX entraîne, grâce à la dénervation partielle du muscle gastrocnémien, une modification de la position de la cheville et par conséquent une amélioration importante de la démarche.
Après injection dans le détrusor, BOTOX inhibe le message efférent contrôlant l'activité motrice du détrusor en bloquant la libération d'acétylcholine. De plus, BOTOX inhibe les neurotransmetteurs afférents et les voies sensitives.
Efficacité clinique
AFFECTIONS NEUROLOGIQUES:
Blépharospasme/hémispasme facial
Au cours d'une étude ouverte, 56 patients souffrant d'hémispasme facial ont reçu une injection initiale de 10 à 50 unités de BOTOX, ils ont été ensuite observés pendant 22 semaines. Les patients sans amélioration à la semaine 4 ont reçu une nouvelle injection (5 à 50 unités). Les patients ont été contrôlés aux semaines 2, 4 (si un nouveau traitement s'avérait nécessaire), 6, 14 et 22. A chaque contrôle, l'effet clinique a été évalué pour les divers muscles inférieurs et supérieurs du visage.
Une amélioration est intervenue chez tous les 56 patients et elle était nette chez 62,5% (35/56) d'entre eux. Une amélioration des muscles supérieurs du visage a été observée chez tous les patients. Lors de l'évaluation des muscles inférieurs du visage, il est apparu que tous les patients sauf deux avaient répondu au traitement.
Une étude supplémentaire, randomisée et multicentrique à double insu avec groupes parallèles, a été effectuée chez des patients avec blépharospasme essentiel bénin pour comparer la sécurité et l'efficacité de deux formulations de BOTOX (la substance active provenait de diverses banques de cellules primaires - dont l'une correspond à la formulation actuelle de BOTOX). Les participants à l'étude avaient déjà été traités par BOTOX. Le traitement consistait en un cycle unique d'injections dans les muscles orbiculaires des deux yeux. Le médecin examinateur déterminait la dose (maximum admissible: 100 unités) et les sites d'injection en fonction de la réponse du patient aux précédents traitements au BOTOX. Les patients sont restés sous observation pendant 12 semaines après le traitement. L'étude comprenait 98 patients (48 dans le groupe traité par la formulation BOTOX actuelle).
Le taux de succès du traitement a été noté de 0 à 4 sur une échelle de 5 points. Le score 0 signifiait «aucun» spasme des paupières et le 4 un spasme «sérieux» invalidant pouvant éventuellement affecter d'autres muscles du visage. En cas de diminution du score ≤2 aux deux yeux, le traitement était considéré comme un succès. Le critère primaire d'évaluation se situait à la semaine 4. Les patients ont reçu une dose moyenne de 33 unités par œil, injectée dans 3 à 15 sites. Le taux de succès de la formulation BOTOX actuelle s'est élevé à environ 90% à la semaine 4, ce qui est comparable avec l'ancienne formulation BOTOX.
Dystonie cervicale
Une étude multicentrique randomisée à double insu et contrôlée contre placebo a été effectuée auprès de patients adultes souffrant de dystonie cervicale (DC) qui avaient répondu positivement à un traitement antérieur par BOTOX. Cette étude comprenait 2 phases de traitement, à savoir une phase ouverte où tous les patients étaient traités par BOTOX, le dosage et les sites d'injection étant déterminés individuellement par le médecin examinateur, et une phase à double insu dans laquelle BOTOX ou le placebo étaient administrés à tous les patients ayant répondu durant la phase ouverte. Un intervalle de jusqu'à 6 semaines séparait ces deux phases. Les patients ont reçu jusqu'à 360 unités BOTOX au total.
Dans la phase d'étude ouverte, 214 patients ont été évalués, dont 170 ont participé à la phase d'étude randomisée en double insu, soit 88 dans le groupe BOTOX et 82 dans le groupe placebo. Par la suite, les patients ont été contrôlés toutes les 2 semaines pendant une période allant jusqu'à 10 semaines après le traitement. Les variables primaires d'efficacité étaient la modification de la position de la tête (mesurée sur l'échelle d'évaluation de la sévérité de la DC) et le pourcentage des patients présentant une amélioration de leur DC, mesurée six semaines après le traitement au moyen d'une échelle d'évaluation médicale. Les douleurs, symptôme important de la DC, ont été évaluées par les patients quant à leur fréquence et leur intensité. En outre, médecins et patients ont apprécié le degré de handicap fonctionnel.
BOTOX s'est avéré significativement supérieur au placebo après 6 semaines selon les deux variables primaires d'efficacité, tant cliniquement que statistiquement. BOTOX a également permis une diminution significativement meilleure au niveau statistique de l'intensité et de la fréquence des douleurs que le placebo. Des améliorations significatives ont aussi été observées à la semaine 6 lors de l'évaluation du handicap fonctionnel par le médecin et le patient.
Une étude supplémentaire, randomisée à double insu en essai croisé, a été effectuée chez des patients souffrant de dystonie cervicale pour analyser la sécurité et l'efficacité de deux formulations de BOTOX (la substance active provenait de diverses banques de cellules primaires – dont l'une correspond à la formulation actuelle de BOTOX). Les participants à l'étude avaient déjà été traités par BOTOX avec des résultats satisfaisants; le traitement comprenait 2 injections séparées par une période de «lavage» de 8 à 16 semaines. Après chaque traitement, les patients ont été observé pendant 8 à 16 semaines, le critère primaire d'évaluation étant déterminé à la semaine 6 après chaque traitement.
Cette étude comprenait 135 patients. La variable primaire d'efficacité était l'évaluation globale de la gravité sur la «Toronto Western Spasmodic Torticollis Rating Scale» (TWSTRS). Les variables secondaires d'efficacité étaient le score d'infirmité/douleur selon la TWSTRS modifiée et l'évaluation globale de la dystonie cervicale par le médecin et le patient. Les résultats ont confirmé qu'une amélioration clinique maximale après l'injection de BOTOX a été atteinte à la semaine 6. La diminution moyenne du score total sur la TWSTRS a correspondu à une amélioration de 35% par rapport aux valeurs initiales; la plus forte diminution moyenne du score d'infirmité/douleurs a correspondu à une amélioration de 50% par rapport à la valeur initiale à ce moment.
L'évaluation globale faite par les médecins et les patients a indiqué également un effet positif du traitement avec un taux de succès noté à la semaine 6 pour la nouvelle formulation de BOTOX de plus de 85% resp. 80% des cas.
Spasticités focales des membres supérieurs chez l'adulte
Une étude multicentrique à double insu et contrôlée contre placebo concernant l'efficacité et la sécurité de BOTOX a été réalisée chez des patients avec spasticité des membres supérieurs suivant un AVC. Les patients étaient des adultes n'ayant jamais été traités par BOTOX, dont l'AVC remontait à ≥6 semaines et qui présentaient des scores d'échelle Ashworth élargie (EAE) de ≥3 au poignet et ≥2 au coude. 91 patients ont été randomisés en quatre groupes de traitement (BOTOX 90, 180 ou 360 unités, ou placebo). Le critère primaire d'évaluation, soit la spasticité du fléchisseur du carpe, a été mesuré à la semaine 6 sur l'échelle EAE. Les variables secondaires d'efficacité étaient la spasticité des fléchisseurs au coude et aux doigts, mesurée sur l'EAE et selon l'évaluation globale du médecin.
L'étude comprenait 91 patients, dont 65 reçurent BOTOX à raison de 90, 180 ou 360 unités. La diminution moyenne du score EAE des fléchisseurs du poignet s'est avérée à la semaine 12 supérieure dans tous les groupes BOTOX à celle du groupe placebo.
Dans tous les groupes de traitement par BOTOX, le tonus du fléchisseur du carpe, mesuré sur l'EAE, a été plus efficacement diminué que sous placebo. Les fléchisseurs du coude ont présenté un type d'amélioration analogue. Concernant les fléchisseurs des doigts, une certaine variabilité a été globalement observée (améliorations significatives seulement dans les semaines 1 et 3), mais les données indiquent des relations de dose/efficacité comparables. L'échelle d'évaluation médicale montre également un avantage significatif des doses de 180 et 360 unités.
Une autre étude multicentrique à double insu et contrôlée contre placebo a été réalisée auprès de patients souffrant de spasticité du poignet et des doigts en tant que conséquence secondaire d'un AVC. La durée de l'étude était de 12 semaines. Le critère primaire d'évaluation était la spasticité du fléchisseur du carpe à la semaine 6, mesurée sur l'échelle d'Ashworth (EA). D'autres variables étaient l'échelle d'évaluation médicale, l'évaluation globale par la personne prenant soin du patient, l'échelle d'évaluation de l'invalidité et les objectifs principaux d'intervention thérapeutique.
Cette étude comprenait 126 patients randomisés et recevant BOTOX (n = 64) ou un placebo (n = 62). La dose de BOTOX injectée se situait entre 200 et 240 unités, répartie entre les muscles atteints du poignet et des doigts. Il est apparu que BOTOX avait amélioré le score EA du poignet et des doigts lors de tous les contrôles après injection. A la semaine 6, 88% des patients présentaient une amélioration sur l'EA sous BOTOX contre 34% dans le groupe placebo.
A la semaine 6, toutes les autres variables présentaient également un avantage significatif par rapport au placebo.
Spasticité focale des membres supérieurs chez les adolescents et les enfants à partir de 2 ans
L'efficacité et l'innocuité de BOTOX pour le traitement de la spasticité des membres supérieurs ont été évaluées chez des patients pédiatriques à partir de 2 ans dans le cadre d'une étude randomisée, multicentrique, en double aveugle, contrôlée par placebo. 234 patients pédiatriques (77 BOTOX 6 unités/kg de poids corporel, 78 BOTOX 3 unités/kg de poids corporel, 79 placebo) souffrant de spasticité due à une «paralysie cérébrale ou un AVC» (score AS modifié pour le coude ou le poignet d'au moins 2) ont été inclus dans l'étude. La dose totale de 3 unités/kg de poids corporel (maximum 100 unités) ou 6 unités/kg de poids corporel (maximum 200 unités) ou placebo a été injectée par voie intramusculaire, répartie entre les muscles du coude ou du poignet et des doigts. Les patients ont été suivis pendant 12 semaines après l'injection. L'utilisation d'un contrôle par électromyogramme (EMG), d'une stimulation nerveuse ou d'une technologie à ultrasons était nécessaire pour aider à localiser correctement le muscle à injecter.
Le critère d'évaluation primaire était la variation moyenne par rapport à la valeur initiale du score AS modifié (ASM) des principaux groupes musculaires (coude ou poignet) au cours des semaines 4 et 6. Le principal critère d'évaluation secondaire était le score moyen de la «Clinical Global Impression of Overall Change by Physician (CGI)» au cours des semaines 4 et 6. La «goal attainment scale» (GAS) évaluée par le médecin pour les objectifs actifs et passifs a été évaluée comme critère secondaire au cours des semaines 8 et 12.
Les patients éligibles pouvaient participer à une étude étendue ouverte dans laquelle ils recevaient plus de 5 traitements à des doses allant jusqu'à 10 unités/kg (maximum 340 unités) si plus d'un membre était traité.
Une amélioration significative par rapport au placebo du score ASM pour la cheville par rapport à la valeur initiale a été observée à tous les moments pour les patients traités par BOTOX dans les deux groupes de traitement. Des résultats similaires ont été observés dans les deux groupes de dose de BOTOX avec une proportion statistiquement significative de répondeurs ASM (définis comme une amélioration d'au moins 1 point) par rapport au placebo dans la plupart des moments.
Tableau 2: Résultats des critères primaires et secondaires principaux
|
|
BOTOX
3 unités/kg
(n=78)
|
BOTOX
6 unités/kg
(n=77)
|
Placebo
(n=79)
| |
Changement moyen par rapport à la valeur initiale dans le groupe musculaire principal (coude ou poignet) sur l'échelle d'Ashworth modifiée**
| |
Moyenne des semaines 4 et 6
|
-1,92*
|
-1,87*
|
-1 21
| |
Clinical Global Impression Score moyen***
| |
Moyenne des semaines 4 et 6
|
1,88
|
1,87
|
1,66
|
* Significativement différent du placebo (p< 0.05)
** L'ASM est une échelle en 6 points (0 [pas d'augmentation du tonus musculaire], 1, 1+, 2, 3 et 4 [membre rigide en flexion ou en extension]), qui mesure la force nécessaire pour déplacer un membre autour d'une articulation, une diminution du score représentant une amélioration de la spasticité.
*** Le CGI évalue la réponse au traitement en termes de qualité de vie du patient sur une échelle de 9 points (-4 = aggravation très significative à +4 = amélioration très significative).
Bien que les valeurs CGI pour BOTOX aient été numériquement plus élevées que pour le placebo, la différence n'était pas statistiquement significative.
Des améliorations statistiquement significatives par rapport au placebo ont été observées dans la proportion de répondeurs CGI (définie comme une amélioration d'au moins 1 point) pour BOTOX 3 unités/kg (semaines 4 à 12) et BOTOX 6 unités/kg (semaine 6), et dans l'atteinte des objectifs fonctionnels (GAS par le médecin) pour les objectifs passifs uniquement pour BOTOX 6 unités/kg (semaine 12).
Figure 6: score ASM (spasticité des membres supérieurs chez les patients pédiatriques) - Changement moyen par rapport à la valeur initiale
Spasticité focale des membres inférieurs chez l'adulte
L'efficacité et l'innocuité de BOTOX pour le traitement de la spasticité des membres inférieurs ont été évaluées dans le cadre d'une étude randomisée, multicentrique, en double aveugle, contrôlée par placebo, qui a inclus 468 patients suite à un AVC (233 BOTOX et 235 placebo) présentant une spasticité de la cheville (score ASM de la cheville d'au moins 3) et dont l'AVC s'est produit au moins 3 mois auparavant. Pour l'étude, 300 à 400 unités de BOTOX ou un placebo ont été injectées par voie intramusculaire dans les muscles M. gastrocnemius, M. soleus et M. tibialis posterior, ainsi que dans des muscles facultatifs, notamment les M. flexor hallucis longus, M. flexor digitorum longus, M. flexor digitorum brevis, M. extensor hallucis et M. rectus femoris. L'utilisation d'un contrôle par électromyogramme (EMG), d'une stimulation nerveuse ou d'une technologie à ultrasons était nécessaire pour aider à localiser correctement le muscle à injecter. Les patients ont été suivis pendant 12 semaines.
Le critère primaire d'évaluation était la variation moyenne du score AMS pour la cheville par rapport à la valeur initiale aux semaines 4 et 6. Un critère secondaire important était le score moyen CGI (évaluation par le médecin de l'impression clinique globale) aux semaines 4 et 6. Des différences statistiquement et cliniquement significatives entre les groupes pour BOTOX par rapport au placebo ont été montrées pour le paramètre d'efficacité primaire ASM et le paramètre clé secondaire CGI et sont présentées dans le tableau ci-dessous. En ce qui concerne le critère primaire d'évaluation du score moyen ASM pour la cheville aux semaines 4 et 6, aucune amélioration par rapport à la valeur initiale n'a été observée chez les patients âgés de 65 ans et plus dans le groupe BOTOX par rapport au placebo, probablement en raison du petit nombre de patients.
Tableau 3: Points d'efficacité primaires et secondaires principaux:
|
|
BOTOX
300 à 400 unités (n=233)
|
Placebo
(n=235)
| |
Changement moyen des fléchisseurs plantaires de la cheville par rapport à la valeur initiale sur l'échelle AMS
| |
Moyenne pour les semaines 4 et 6
|
-0,8*
|
-0,6
| |
Valeur moyenne de l'impression clinique globale du médecin investigateur
| |
Moyenne pour les semaines 4 et 6
|
0,9*
|
0,7
| |
Changement moyen des fléchisseurs des orteils sur l'échelle AMS
| |
FHL** Moyenne pour les semaines 4 et 6
|
-1,0*
|
-0,6
| |
FDL + FDB** Moyenne pour les semaines 4 et 6
|
-0,9
|
-0,8
|
* Significativement différent du placebo (p<0,05)
FHL = flexor hallucis longus; FDL = flexor digitorum longus; FDB = flexor digitorum brevis
Figure 7: Score AMS (spasticité des membres inférieurs chez l'adulte) - Changement moyen par rapport à la valeur initiale
Figure 8: CGI du médecin (spasticité des membres inférieurs chez l'adulte) - valeurs moyennes par visite
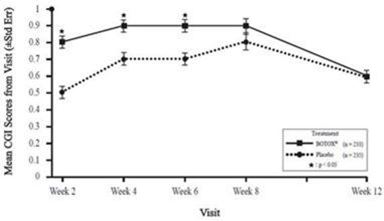
Spasticité focale des membres inférieurs chez les adolescents et les enfants à partir de 2 ans
L'efficacité et l'innocuité de BOTOX pour le traitement de la spasticité des membres inférieurs chez les patients pédiatriques à partir de 2 ans ont été évaluées dans le cadre d'une étude randomisée, multicentrique, en double aveugle, contrôlée par placebo. L'étude a porté sur 384 patients pédiatriques (128 BOTOX 8 unités/kg, 126 BOTOX 4 unités/kg et 128 placebo) présentant une spasticité des membres inférieurs due à une infirmité motrice cérébrale (score ASM pour la cheville d'au moins 2). Une dose totale de 4 unités/kg (maximum 150 unités) ou 8 unités/kg (maximum 300 unités) ou de placebo a été injectée par voie intramusculaire, répartie entre le gastrocnémien, le soleus et le tibialis posterior. L'utilisation d'un contrôle par électromyogramme (EMG), d'une stimulation nerveuse ou d'une technologie à ultrasons était nécessaire pour aider à localiser correctement le muscle à injecter. Les patients ont été suivis pendant 12 semaines. Le critère primaire d'évaluation était le changement moyen par rapport à la valeur initiale du score ASM pour la cheville lors des semaines 4 et 6, et le critère d'évaluation secondaire principal était le CGI moyen lors des semaines 4 et 6. Les critères d'évaluation secondaires lors des semaines 8 et 12 comprenaient le GAS par le médecin et la Edinburgh Visual Gait (EVG) (chez un sous-ensemble de patients).
Les patients éligibles pouvaient participer à une étude étendue ouverte dans laquelle ils recevaient jusqu'à cinq traitements à des doses allant jusqu'à 10 unités/kg (maximum 340 unités) avec traitement de plus d'un membre.
Une amélioration statistiquement significative du critère primaire par rapport au placebo a été observée chez les patients traités avec 4 et 8 unités/kg de BOTOX pendant la semaine 12 (voir graphique 4). Des résultats similaires ont été observés dans les deux groupes de dose de BOTOX avec une proportion plus élevée de répondeurs ASM (définis comme une amélioration d'au moins 1 point) par rapport au placebo dans la plupart des moments.
Tableau 4: Résultats des critères d'efficacité primaires et secondaires principaux
|
|
BOTOX 4 unités/kg
(n = 125)
|
BOTOX 8 unités/kg
(n=127)
|
Placebo
(n=129)
| |
Changement moyen de la valeur initiale ASM des fléchisseurs plantaires
| |
Moyenne semaines 4 et 6
|
-1,01*
|
-1,06*
|
-0 80
| |
Valeur moyenne CGI
| |
Moyenne semaines 4 et 6
|
1,49
|
1,65*
|
1,36
|
* Différence significative par rapport au placebo (p< 0,05)
Une amélioration statistiquement significative par rapport au placebo a été démontrée avec 4 et 8 unités/kg de BOTOX pour le critère primaire d'évaluation du score ASM pour la cheville lors des semaines 4 et 6, en tant que changement par rapport à la valeur initiale, et avec 8 unités/kg de BOTOX pour le critère secondaire principal, le CGI moyen lors des semaines 4 et 6. Une amélioration statistiquement significative par rapport au placebo a été observée pour le CGI par le médecin sur une moyenne de 4 et 6 semaines pour BOTOX 8 unités/kg. Des améliorations statistiquement significatives de la réalisation des objectifs fonctionnels actifs et passifs (GAS) ont été observées lors des semaines 8 et 12 pour BOTOX 8 unités/kg et lors de la semaine 8 mais pas lors de la semaine 12 pour BOTOX 4 unités/kg. L'amélioration de la marche dépendait de la dose, avec des améliorations statistiquement significatives de l'EVG à la semaine 8 pour BOTOX 8 unités/kg.
Figure 9: score ASM (spasticité des membres inférieurs chez les patients pédiatriques) - Changement moyen par rapport à la valeur initiale
Chez les patients pédiatriques atteints de spasticité des membres inférieurs dont les échantillons ont été analysés dans le cadre d'une étude de phase 3 et de l'étude Open Label, 2 patients sur 264 (0,8%) ont développé des anticorps neutralisants lorsqu'ils ont été traités par BOTOX pendant un maximum de 5 cycles de traitement. Les deux patients ont ensuite continué à bénéficier des avantages cliniques du traitement par BOTOX.
Prophylaxie des céphalées chez les patients adultes atteints de migraine chronique
BOTOX a été évalué dans le cadre de deux études multinationales multicentriques de 56 semaines, qui comprenaient les éléments suivants: une phase en double aveugle de 24 semaines, avec 2 cycles d'injections comparant BOTOX à un placebo, suivie d'une phase en ouvert de 32 semaines, avec 3 cycles d'injection de BOTOX. Au total, 1 384 adultes atteints de migraine chronique qui n'avaient encore jamais reçu de traitement prophylactique concomitant pour les céphalées, ou qui ne l'avaient pas pris au cours d'une période de 28 jours avant le début de l'étude, et qui ont ≥15 jours de céphalées, dont 50 % migraine/migraine probable et ≥4 épisodes de céphalées, ont été évalués dans le cadre de deux études cliniques de phase 3. Ces patients ont été randomisés pour recevoir, toutes les 12 semaines, des injections avec un placebo ou 155 à 195 unités de BOTOX pour la phase en double aveugle avec 2 cycles d'injections. Ils pouvaient recevoir 3 cycles de BOTOX supplémentaires en ouvert, avec un total de 5 cycles d'injections maximum
Le traitement aigu des maux de tête était autorisé pour les patients (65,5 % d'entre eux avaient reçu un traitement aigu excessif pendant la phase de démarrage de l'étude).
Le traitement par BOTOX a montré, par rapport au placebo, une amélioration statistiquement significative (p < 0,001) et cliniquement pertinente par rapport à la valeur initiale en ce qui concerne la fréquence des jours de céphalées, la fréquence des jours de migraine/migraine probable, la fréquence des jours de céphalées modérées/sévères et le nombre total d'heures cumulées de céphalées les jours de céphalées.
Les résultats du test Headache Impact Test (HIT6) et du questionnaire de qualité de vie spécifique à la migraine (MSQ) ont indiqué une efficacité durable de BOTOX ainsi qu'une amélioration des capacités fonctionnelles, de la vitalité, de la charge psychique et globalement, de la qualité de vie.
Résultats groupés des principaux critères d'évaluation d'efficacité lors des deux études pivots réalisées la semaine 24 (critère d'évaluation principal)
|
Efficacité sur 28 jours
|
BOTOX
(N = 688)
|
Placebo
(N = 696)
|
Valeur p
| |
Variation du nombre de jours avec céphalées par rapport à la valeur initiale
|
-8,4
|
-6,6
|
< 0,001
| |
Variation du nombre de jours de migraine/de migraine probable par rapport à la valeur initiale
|
-8,2
|
-6,2
|
< 0,001
| |
Variation du nombre de jours avec céphalées modérées/sévères par rapport à la valeur initiale
|
-7,7
|
-5,8
|
< 0,001
| |
Variation du nombre total d'heures avec céphalées pendant les jours avec céphalées par rapport à la valeur initiale
|
-119,7
|
-80,5
|
< 0,001
| |
Variation du nombre d'épisodes de céphalées par rapport à la valeur initiale
|
-5,2
|
-4,9
|
0,009
| |
Pourcentage de patients ayant au moins 50 % de céphalées en moins
|
47%
|
35%
|
< 0,001
| |
Proportion de patients ayant un score élevé dans les catégories de l'HIT-6
|
67,6 %
|
78,2 %
|
< 0,001
| |
Variation du score total HIT-6 par rapport à la valeur initiale
|
-4,8
|
-2,4
|
< 0,001
|
Bien que les études n'aient pas été conçues pour indiquer des différences concernant les sous-groupes, l'effet du traitement a semblé plus faible dans le sous-groupe des participants de sexe masculin (n = 188) et des participants qui n'étaient pas d'origine caucasienne (n = 137), par rapport à la population totale de l'étude.
AFFECTIONS VÉSICALES:
Vessie hyperactive chez les adultes
Deux études cliniques de phase 3, multicentriques, randomisées, contrôlées contre placebo, menées en double aveugle sur une période de 24 semaines ont été effectuées auprès de patients atteints de vessie hyperactive accompagnée des symptômes suivants: incontinence urinaire, mictions impérieuses et mictions fréquentes. Au total, 1'105 patients dont les symptômes n'ont pas pu être contrôlés de façon adéquate par les anticholinergiques (réponse insuffisante ou effets secondaires inacceptables) ont été randomisés pour recevoir soit 100 unités BOTOX (n = 557) ou un placebo (n = 548).
Dans les deux études, des améliorations significatives par rapport au placebo ont été constatées pour BOTOX (100 unités) en ce qui concerne la modification de la fréquence journalière des épisodes d'incontinence urinaire par rapport à la valeur initiale, y compris la proportion de patients continents, à la semaine 12 (critère primaire). La proportion des patients, mesurée au moyen de l'échelle pour le bénéfice du traitement, qui ont rapporté une réponse positive au traitement (leur état s'était «fortement amélioré» ou «amélioré») était, dans les deux études, significativement plus élevée dans le groupe BOTOX que dans le groupe placebo.
Par rapport au placebo, des améliorations significatives ont également été observées au niveau de la fréquence mictionnelle journalière et de la fréquence des épisodes de mictions impérieuses et de nycturie. La quantité d'urine excrétée par miction était également significativement plus élevée. Des améliorations significatives ont été constatées à partir de la semaine 2 pour tous les symptômes de la vessie hyperactive.
Par comparaison avec le placebo, le traitement par BOTOX a été associé à des améliorations significatives de la qualité de vie liée à la santé, mesurée au moyen du questionnaire I-QOL (Incontinence Quality of Life) (avec les facteurs suivants: comportement d'éviction et restrictions, conséquences psychosociales et gêne sociale) et du questionnaire (King's Health Questionnaire) (avec les facteurs suivants: conséquences de l'incontinence, limitations des activités quotidiennes, limitations sociales, limitations corporelles, relations personnelles, état émotionnel, sommeil/énergie et degré de sévérité/contre-mesures).
Dans l'ensemble, aucune différence au niveau de l'effet n'a été constatée entre les patients de 65 ans et plus et les patients de moins de 65 ans suite au traitement par BOTOX.
Les résultats des études centrales compilées sont représentés ci-dessous:
Tableau 5: Critères primaires et secondaires au début de l'étude et modification par rapport au début de l'étude dans les études centrales compilées
|
|
BOTOX 100
unités
(n = 557)
|
Placebo
(n = 548)
|
Valeur p
| |
Fréquence journalière des épisodes d'incontinence urinaire*
| |
Valeur moyenne au début de l'étude
|
5,49
|
5,39
|
| |
Modification moyenne à la semaine 2
|
-2,85
|
-1,21
|
<0,001
| |
Modification moyenne à la semaine 6
|
-3,11
|
-1,22
|
<0,001
| |
Modification moyenne à la semaine 12
|
-2,80
|
-0,95
|
<0,001
| |
Proportion de réponse positive au traitement sur l'échelle du bénéfice du traitement (%)
| |
Semaine 2
|
64,4
|
34,7
|
<0,001
| |
Semaine 6
|
68,1
|
32,8
|
<0,001
| |
Semaine 12
|
61,8
|
28,0
|
<0,001
| |
Fréquence mictionnelle journalière
| |
Valeur moyenne au début de l'étude
|
11,99
|
11,48
|
| |
Modification moyenne à la semaine 2
|
-1,53
|
-0,78
|
<0,001
| |
Modification moyenne à la semaine 6
|
-2,18
|
-0,97
|
<0,001
| |
Modification moyenne à la semaine 12
|
-2,35
|
-0,87
|
<0,001
| |
Fréquence journalière des épisodes de mictions impérieuses
| |
Valeur moyenne au début de l'étude
|
8,82
|
8,31
|
| |
Modification moyenne à la semaine 2
|
-2,89
|
-1,35
|
<0,001
| |
Modification moyenne à la semaine 6
|
-3,56
|
-1,40
|
<0,001
| |
Modification moyenne à la semaine 12b
|
-3,30
|
-1,23
|
<0,001
| |
Score total I-QOL
| |
Valeur moyenne au début de l'étude
|
34,1
|
34,7
|
| |
Modification moyenne après la semaine 12bc
|
+22,5
|
+6,6
|
<0,001
| |
Questionnaire KHQ: Limitation des activités
| |
Valeur moyenne au début de l'étude
|
65,4
|
61,2
|
| |
Modification moyenne après la semaine 12bc
|
-25,4
|
-3,7
|
<0,001
| |
Questionnaire KHQ: limitations sociales
| |
Valeur moyenne au début de l'étude
|
44,8
|
42,4
|
<0,001
| |
Modification moyenne après la semaine 12bc
|
-16,8
|
-2,5
|
<0,001
|
* Dans le groupe BOTOX, la proportion de patients continents à la semaine 12 était de 27,1% et de 8,4% dans le groupe placebo. La proportion des patients atteignant une fréquence réduite d'au moins 75% ou 50% des épisodes d'incontinence par rapport à la valeur initiale était de 46,0 % ou 60,5 % dans le groupe BOTOX et de 17,7% ou 31,0% dans le groupe placebo.
a Critères d'évaluation coprimaires
b Critères d'évaluation secondaires
c La différence minimale significative prédéfinie par rapport à la valeur initiale était de +10 points pour le I-QOL et de -5 points pour le KHQ.
La durée médiane de la réponse suite au traitement par BOTOX, basée sur une demande de retraitement exprimée par les patients, était de 166 jours (~24 semaines).
Chez les patients qui ont également participé à l'étude de prolongation ouverte et qui ont été traités uniquement avec 100 unités de BOTOX (n = 438), la durée médiane de la réponse était de 212 jours (~30 semaines).
Seul un nombre limité de patients de sexe masculin (12,2% de la population totale de l'étude) ont pris part aux deux études pivots. Un effet statistiquement significatif n'a pas pu être mis en évidence dans ce petit sous-groupe.
Au total, 834 patients ont été examinés dans une étude de prolongation au long cours. Lors de nouveaux traitements, les patients ont montré une réponse constante pour tous les critères d'évaluation de l'efficacité.
Dans les études centrales, aucun des 615 patients chez lesquels des échantillons ont été analysés n'a développé d'anticorps neutralisants. Chez les patients dont les échantillons issus de l'étude pivot de phase 3 et des études de prolongation ouvertes ont été analysés, des anticorps neutralisants se sont développés dans les cas suivants: chez 0 patient sur 954 (0,0%) durant le traitement avec 100 unités de BOTOX et chez 3 patients sur 260 (1,2%) après un traitement ultérieur avec au minimum une dose de 150 unités. L'un de ces trois patients a présenté un bénéfice clinique durable.
Hyperactivité vésicale chez les enfants et adolescents
Des données à efficacité limitée sont issues d'une étude randomisée multi-centrique en double aveugle à groupes parallèles (191622 137), menée auprès de patients âgés de 12 à 17 ans présentant une hyperactivité vésicale et des symptômes d'incontinence urinaire. Les patients n'ayant pas répondu de manière adéquate ou étant intolérants à au moins un médicament anti-cholinergique ont été inclus dans cette étude. En raison de problèmes de recrutement, seulement 55 des 108 patients prévus ont été inclus, ce qui signifie que le nombre de cas était insuffisant pour pouvoir se prononcer sur une efficacité pour cette population. Ces patients ont été randomisés pour recevoir 25 unités, 50 unités ou 100 unités, la dose de 6 unités/kg de poids corporel ne devant pas être dépassée; BOTOX 25 unités N = 18, BOTOX 50 unités N = 17 et BOTOX 100 unités N = 20. Avant utilisation, les patients ont reçu une anesthésie conformément à la pratique locale. Tous les patients ont reçu une anesthésie générale ou une légère sédation.
En ce qui concerne le critère d'évaluation principal à la semaine 12, «Fréquence quotidienne des épisodes d'incontinence urinaire diurne», ainsi que d'autres critères tels que la fréquence des épisodes quotidiens de miction, la fréquence des épisodes quotidiens de besoin impérieux d'uriner ou une réponse positive au traitement, des différences numériques sans aucune différence statistiquement significative ont pu parfois être constatées entre les groupes de traitement.
Sur les 55 patients pédiatriques qui présentaient une valeur initiale négative pour les anticorps de liaison ou les anticorps neutralisants, et après le début de l'étude au moins une valeur évaluable par rapport à la valeur initiale issue d'une étude randomisée en double aveugle, aucun n'a développé d'anticorps neutralisants après traitement avec 25–100 unités de BOTOX.
Incontinence urinaire chez les adultes à la suite d'une hyperactivité neurogène du détrusor
Deux études cliniques de phase 3, multicentriques, randomisées, en double aveugle, contrôlées contre placebo, ont été réalisées chez des patients présentant une incontinence urinaire due à une hyperactivité neurogène du détrusor, soit avec miction spontanée, soit sondés. Au total, 691 patients présentant une lésion médullaire ou atteints de sclérose en plaques et qui n'étaient pas traités d'une manière satisfaisante par au moins un agent anticholinergique ont participé aux études. Les patients ont été randomisés pour recevoir soit 200 unités de BOTOX (n = 227), soit 300 unités de BOTOX (n = 223), soit un placebo (n = 241).
Dans les deux études de phase 3, des améliorations significatives ont été observées par rapport au placebo concernant le critère principal d'efficacité (modification de la fréquence hebdomadaire des épisodes d'incontinence, par rapport au début de l'étude): de meilleurs résultats ont été notés sous BOTOX (200 unités et 300 unités) à la semaine 6 (temps d'évaluation du critère d'efficacité) y compris pour les patients chez lesquels l'incontinence urinaire avait disparu. Des améliorations significatives des paramètres urodynamiques ont également été observées, y compris une augmentation de la capacité cystométrique maximale et une diminution du pic de pression sur le muscle détrusor durant la première contraction involontaire de ce dernier. Par rapport au placebo, on a également observé des améliorations significatives de l'évaluation de la qualité de vie par le patient spécifiquement en rapport avec l'incontinence et la santé, déterminées au moyen d'un questionnaire I-QOL (incluant tactiques d'évitement, conséquences psychosociales et sentiments de honte restrictifs). Un bénéfice supplémentaire de BOTOX 300 unités par rapport à 200 unités n'a pas pu être mis en évidence.
Les résultats compilés des études pivots sont présentés ci-dessous:
Tableau 6: Résultats compilés des études pivots concernant les critères d'évaluation primaires et secondaires au début de l'étude et les changements par rapport au début de l'étude
|
|
BOTOX
200 unités
(n = 227)
|
Placebo
(n = 241)
|
Valeur p
| |
Fréquence hebdomadaire de l'incontinence urinaire*
| |
Valeur moyenne au début de l'étude
|
32,4
|
31,5
|
| |
Modification moyenne à la semaine 2
|
-17,7
|
-9,0
|
<0,001
| |
Modification moyenne à la semaine 6a
|
-21,3
|
-10,5
|
<0,001
| |
Modification moyenne à la semaine 12
|
-20,6
|
-9,9
|
<0,001
| |
Capacité cystométrique maximale (ml)
| |
Valeur moyenne au début de l'étude
|
250,2
|
253,5
|
| |
Modification moyenne à la semaine 6b
|
+153,6
|
+11,9
|
<0,001
| |
Pression maximale détrusorienne durant la première contraction involontaire du détrusor (cmH2O):
| |
Valeur moyenne au début de l'étude
|
51,5
|
47,3
|
| |
Modification moyenne à la semaine 6b
|
-32,4
|
+1,1
|
<0,001
| |
Score global relatif à la qualité de vie spécifiquement en rapport avec l'incontinencec,d
| |
Valeur moyenne au début de l'étude
|
35,37
|
35,32
|
| |
Modification moyenne à la semaine 6b
|
+25,89
|
+11,15
|
<0,001
| |
Modification moyenne à la semaine 12
|
+28,89
|
+8,86
|
<0,001
|
* Dans le groupe traité par 200 unités de BOTOX, la proportion de patients continents était de 37% pendant toute la semaine 6 et de 9% dans le groupe sous placebo. La proportion atteignant une réduction de 75% au moins de la fréquence des épisodes d'incontinence par rapport au début de l'étude était de 63% et 24%. La proportion atteignant une réduction de 50% au moins de la fréquence des épisodes d'incontinence par rapport au début de l'étude était de 76% et 39%.
a Critère d'évaluation primaire
b Critère d'évaluation secondaire
c L'échelle du score total de l'I-QOL va de 0 (problème majeur) à 100 (absolument aucun problème).
d Dans les études pivots, la différence minimale significative prédéfinie (MID – Minimally Important Difference) du score global I-QOL était de 8 points, en partant d'une valeur estimée de MID de 4 à 11 points indiquée par les patients avec une hyperactivité neurogène du détrusor.
La durée médiane de l'effet du traitement dans les deux études pivots, basée sur une demande de retraitement exprimée par le patient, était de 256 à 295 jours (36 à 42 semaines) pour le groupe ayant reçu 200 unités de BOTOX comparé à 92 jours (13 semaines) pour le groupe placebo. Chez les patients qui ont également participé à l'étude de prolongation ouverte et qui ont été traités uniquement avec 200 unités de BOTOX (n = 174), la durée médiane de la réponse était de 253 jours (~36 semaines).
Lors de la répétition du traitement, la réponse des patients est restée constante concernant tous les critères d'évaluation de l'efficacité.
Dans les études pivots, aucun des 475 patients présentant une hyperactivité neurogène du détrusor et pour lesquels des échantillons ont été analysés n'a développé des anticorps neutralisants.
Chez les patients dont les échantillons issus du programme de développement de nouveaux médicaments (y compris étude de prolongation ouverte) ont été analysés, des anticorps neutralisants se sont développés dans les cas suivants: chez 3 patients sur 300 (1,0%) traités uniquement avec 200 unités de BOTOX et chez 5 patients sur 258 (1,9%) après un traitement avec au minimum une dose de 300 unités. Quatre de ces huit patients ont présenté un bénéfice clinique durable.
Après l'autorisation de mise sur le marché, une étude a été réalisée en double aveugle et contre placebo sur des patients atteints de sclérose en plaques (SEP), lesquels présentaient une incontinence urinaire causée par une hyperactivité neurogène du détrusor et n'ont pas été traités de manière satisfaisante par au moins un agent anticholinergique et n'étaient pas cathétérisés en début d'étude. Ces patients ont été randomisés pour recevoir soit 100 unités de BOTOX (n = 66) soit un placebo (n = 78).
Des améliorations significatives du paramètre d'efficacité primaire, une modification dans la fréquence quotidienne des épisodes d'incontinence par rapport au début de l'étude, ont été observées pour BOTOX (100 unités) à la période d'efficacité primaire en semaine 6 en comparaison avec un placebo. Cela englobait également le groupe de patients chez qui l'incontinence urinaire avait disparu. Des améliorations significatives des paramètres urodynamiques et du questionnaire I-QOL rempli par le patient ont été observées. Cela inclut les tactiques d'évitement, les conséquences psychosociales et les sentiments de honte restrictifs.
Les résultats de l'étude après introduction sur le marché sont présentés comme suit:
Tableau 7: Critères primaires et secondaires au début de l'étude et modification par rapport au début de l'étude après introduction sur le marché avec 100 unités de BOTOX et dans le cas de patients SEP progressive non cathétérisés en début d'étude
|
|
BOTOX
100 unités (n=66)
|
Placebo
(n=78)
|
Valeur p
| |
Fréquence journalière des épisodes d'incontinence urinaire*
| |
Valeur moyenne au début de l'étude
|
4.2
|
4.3
|
| |
Modification moyenne à la semaine 2
|
-2.9
|
-1.2
|
p<0.001
| |
Modification moyenne à la semaine 6a
|
-3.3
|
-1.1
|
p<0.001
| |
Modification moyenne à la semaine 12
|
-2.8
|
-1.1
|
p<0.001
| |
Capacité cystométrique maximale (ml)
| |
Valeur moyenne au début de l'étude
|
246.4
|
245.7
|
| |
Modification moyenne à la semaine 6b
|
+127.2
|
-1.8
|
p<0.001
| |
Pression maximale détrusorienne durant la première contraction involontaire du détrusor (cmH2O):
| |
Valeur moyenne au début de l'étude
|
35.9
|
36.1
|
| |
Modification moyenne à la semaine 6b
|
-19.6
|
+3.7
|
p=0.007
| |
Score global relatif à la qualité de vie spécifiquement en rapport avec l'incontinencec,d
| |
Valeur moyenne au début de l'étude
|
32.4
|
34.2
|
| |
Modification moyenne à la semaine 6b
|
+40.4
|
+9.9
|
p<0.001
| |
Modification moyenne à la semaine 12
|
+38.8
|
+7.6
|
p<0.001
|
* Dans le groupe traité par 100 unités de BOTOX, la proportion de patients continents était de 53% pendant toute la semaine 6 et de 10,3% dans le groupe sous placebo.
a Critère d'évaluation primaire
b Critère d'évaluation secondaire
c L'échelle du score total de l'I-QOL va de 0 (problème majeur) à 100 (absolument aucun problème).
d La différence minimale significative prédéfinie (MID – Minimally Important Difference) du score global I-QOL était de 11 points, en partant d'une valeur estimée de MID de 4 à 11 points indiquée par les patients avec une hyperactivité neurogène du muscle détrusorien.
La durée médiane de la réponse dans cette étude, basée sur une demande de retraitement exprimée par les patients, était de 362 jours (~52 semaines) dans le groupe traité avec 100 unités de BOTOX contre 88 jours (~13 semaines) pour le groupe ayant reçu un placebo.
Hyperactivité neurogène du détrusor chez les patients pédiatriques à partir de 5 ans
Une étude clinique randomisée, multicentrique, en double aveugle avec groupes de doses parallèles, mais sans bras placebo [en raison de la population pédiatrique étudiée] (191622-120) a été réalisée chez des patients entre 5 et 17 ans, présentant une incontinence urinaire à la suite d'une hyperactivité du détrusor associée à une affection neurologique, traités par sondage propre intermittent et ayant répondu de manière insuffisante à une thérapie par anticholinergiques ou ne l'ayant pas toléré. Au total, 113 patients (dont 99 présentant des défauts du tube neural, comme le spina bifida ou le syndrome de la moelle attachée, 13 présentant des lésions de la moelle épinière [T1 ou en-dessous] et 1 présentant une myélite transverse) ont été traités par BOTOX. Ces patients ont reçu de manière aléatoire 50 unités, 100 unités ou 200 unités, sans pouvoir dépasser 6 unités/kg de poids. Les patients qui recevaient moins que la dose à laquelle ils avaient été randomisés en raison de la quantité maximale de 6 unités/kg ont été classés dans le groupe de dose le plus proche pour l'analyse. Au total, 38 patients ont été assignés au groupe recevant 50 unités, 45 patients au groupe recevant 100 unités et 30 patients au groupe recevant 200 unités de BOTOX. Avant le traitement, les patients ont reçu une anesthésie selon leur âge et la pratique locale. 109 patients (97,3 %) ont reçu une anesthésie générale ou une sédation consciente et 3 patients (2,7 %) ont reçu une anesthésie locale.
À la semaine 6 (première date), les résultats de l'étude ne montraient aucune différence statistiquement significative de la première variable d'efficacité [approche statistique prédéfinie], de la modification de la fréquence moyenne des épisodes d'incontinence urinaire quotidienne par rapport à la valeur initiale, entre les groupes 100 unités et 50 unités de BOTOX (p = 0,9949) ou entre les groupes 200 unités et 50 unités de BOTOX (p = 0,9123). Au sein du groupe, des améliorations d'environ 30 % ont été constatées dans tous les groupes de doses en ce qui concerne les épisodes d'incontinence urinaire quotidienne (normalisés à 12 heures).
Pour la deuxième variable d'efficacité, le volume urinaire après le premier sondage matinal, des différences de groupe significatives sont apparues à la semaine 6 entre le groupe de dose traité avec 200 U par rapport à celui traité avec 50 U de BOTOX (p = 0,0055), alors que l'augmentation dans le groupe recevant 100 unités de BOTOX par rapport à 50 unités n'était pas statistiquement significative (p = 0,5117).
En ce qui concerne les mesures pour réduire la pression maximale de la vessie à la semaine 6, l'administration de 200 unités de BOTOX s'est avérée significativement plus efficace que l'administration de 50 unités de BOTOX (p = 0,0157), en revanche, la dose de 100 U non (p = 0,1737).
Tableau 8: Valeur initiale et variation par rapport à la valeur initiale en ce qui concerne la fréquence quotidienne des épisodes d'incontinence urinaire, le volume urinaire lors du premier sondage matinal et la pression maximale du détrusor pendant la phase de stockage (cmH2O) et la capacité cystométrique maximale (ml) dans l'étude 191622-120
|
|
BOTOX 200 unités
n = 30
|
Valeur p*
| |
Fréquence quotidienne moyenne des épisodes d'incontinence urinaire pendant la journéea
| |
Valeur moyenne au début de l'étude
|
3,7
|
| |
Variation moyenne* à la semaine 2 (IC à 95 %)
|
–1,1 (–1,7, –0,6)
|
< 0,0001
| |
Variation moyenne* à la semaine 6** (IC à 95 %)
|
–1,3 (–1,8, –0,9)
|
< 0,0001
| |
Variation moyenne* à la semaine 12 (IC à 95 %)
|
–0,9 (–1,5, –0,4)
|
0,0009
| |
Volume urinaire lors du premier sondage matinal (ml)b
| |
Valeur moyenne au début de l'étude
|
187,7
|
| |
Variation moyenne* à la semaine 2 (IC à 95 %)
|
63,2 (27,9, 98,6)
|
0,0006
| |
Variation moyenne* à la semaine 6** (IC à 95 %)
|
87,5 (52,1, 122,8)
|
< 0,0001
| |
Variation moyenne* à la semaine 12 (IC à 95 %)
|
45,2 (10,0, 80,5)
|
0,0125
| |
Pression maximale du détrusor (Pdetmax) pendant la phase de stockage (cm H2O)b
| |
Valeur moyenne au début de l'étude
|
56,7
|
| |
Variation moyenne* à la semaine 6** (IC à 95 %)
|
–27,3 (–36,4, –18,2)
|
< 0,0001
| |
Capacité cystométrique maximale (ml) (MCC)b
Valeur moyenne au début de l'étude
| |
Variation moyenne* à la semaine 6** (IC à 95 %)
|
202,3
|
| |
Contraction involontaire du détrusor
|
63,6 (29,0, 98,1)
|
| |
Début de l'étude
|
25 (92,6)
|
| |
Semaine 6**
|
13 (46,4)
|
0,0004
|
IC = intervalle de confiance
* La variation de la moyenne des moindres carrés et la valeur p sont basées sur le modèle ANCOVA avec la valeur initiale comme covariable et le groupe de traitement, l'âge (< 12 ans ou ≥12 ans), le nombre d'épisodes d'incontinence urinaire diurne à l'inclusion (≤6 ou > 6) et le traitement anticholinergique (oui/non) à l'inclusion comme facteurs.
**Date principale
a Critère d'évaluation principal
b Critère d'évaluation secondaire
Dans l'ensemble, les données relatives au traitement répété ont montré un bénéfice persistant.
Aucun des 99 patients pédiatriques qui présentaient un taux initial négatif d'anticorps de liaison ou neutralisants au début de l'étude et qui avaient au moins une valeur évaluable au cours d'une étude randomisée en double aveugle et d'une étude d'extension en double aveugle n'a développé d'anticorps neutralisants après avoir reçu jusqu'à quatre cycles de traitement par 50 à 200 unités de BOTOX.
AFFECTIONS DE LA PEAU ET APPERENTÉES:
Hyperhidrose axillaire primaire
Une étude clinique multicentrique à double insu a été réalisée auprès de patients souffrant d'une hyperhidrose axillaire primaire bilatérale persistante. L'hyperhidrose axillaire primaire était présente lorsque la mesure initiale indiquait une production spontanée d'au moins 50 mg de sueur par aisselle en 5 minutes, au repos et à la température ambiante. L'étude comprenait 320 patients randomisés recevant 50 unités de BOTOX (n = 242) ou un placebo (n = 78). La réponse au traitement était définie comme une diminution de la production de sueur d'au moins 50% par rapport à la valeur initiale. Au critère d'évaluation primaire, 4 semaines après l'injection, 93,8% des patients du groupe BOTOX avaient répondu au traitement contre 35,9% du groupe placebo (p < 0,001). Lors de tous les contrôles durant les 16 semaines suivant l'injection, le taux de réponse dans le groupe BOTOX s'est continuellement montré supérieur (p < 0,001) à celui du groupe placebo.
Une étude ouverte a été ensuite réalisée auprès de 207 patients adéquats qui ont reçu jusqu'à 3 traitements par BOTOX. Parmi ceux-ci, 174 ont terminé les 16 mois au total que duraient les deux études combinées (4 mois en double aveugle et 12 mois à la suite en étude ouverte). Le taux de réponse 16 semaines après le premier (n = 287), le deuxième (n = 123) et le troisième (n = 30) traitement s'est élevé à 85,0%, 86,2% et 80%. Au cours des deux études combinées, la durée moyenne de l'effet a été de 7,5 mois après le premier traitement, mais l'effet a persisté encore un an ou plus chez 27,5% des patients.
PharmacocinétiqueDes études classiques portant sur l'absorption, la distribution, la biotransformation et l'élimination de la substance active n'ont pas été réalisées en raison de la nature de la toxine botulique de type A.
Propriétés générales de la substance active
Distribution/Métabolisme/Élimination
Les études portant sur la distribution chez les rats indiquent une lente diffusion musculaire de la toxine botulique A radiomarquée dans le muscle gastrocnémien. Elle est suivie d'une métabolisation systémique rapide et d'une excrétion urinaire.
La demi-vie du produit radiomarqué était d'environ 10 heures. Au point d'injection, les éléments radioactifs étaient liés à de grosses molécules protéiques, tandis que dans le plasma, la radioactivité était liée à de petites molécules. Cela suggère une métabolisation systémique rapide.
Dans les 24 heures après l'administration de la dose, 60% de la radioactivité était excrétée dans les urines. La toxine est probablement métabolisée par des protéases et les composants moléculaires sont recyclés par les voies métaboliques normales.
Distribution
Distribution du principe actif dans le patient:
On pense que la distribution systémique des doses thérapeutiques de BOTOX est faible. Des études cliniques utilisant des techniques électromyographiques à fibre unique ont mis en évidence une augmentation de l'activité électrophysiologique neuromusculaire dans les muscles distants du point d'injection, sans aucun signe ou symptôme clinique associé. L'ampleur de la diffusion et des effets secondaires associés dépend des caractéristiques anatomiques, ainsi que des volumes et de la dose injectés.
Données précliniquesPharmacologie de sécurité
Outre les études de toxicité pour la reproduction, les études de sécurité précliniques suivantes ont été conduites avec BOTOX: toxicité aiguë, toxicité chronique, tolérance locale, potentiel mutagène, pouvoir antigénique, compatibilité sanguine chez l'homme. Ces études n'ont révélé aucun risque particulier chez l'homme aux doses cliniquement significatives.
Aucune toxicité systémique n'a été observée après une injection unique dans le détrusor de <50 unités /kg de BOTOX chez le rat. Afin de simuler une injection accidentelle, une dose unique de BOTOX (~7 unités /kg) a été administrée dans l'urètre prostatique et le rectum proximal, la vésicule séminale et la paroi vésicale ainsi que dans l'utérus (~3 unités /kg) de singes sans qu'aucun effet clinique indésirable n'ait été observé.
Dans une étude d'administration répétée dans le détrusor de singes pendant 9 mois (4 injections), une ptose a été observée à la dose de 24 unités/kg. Des cas de mortalité ont été observés après une dose de 36 unités/kg après la première des quatre injections et chez un animal à la dose de 24 unités/kg après la deuxième injection. On a observé une activité motrice réduite et des difficultés respiratoires chez des animaux moribonds après administrations de doses ≥24 unités/kg. Aucun effet indésirable n'a été observé chez le singe à la dose de 12 unités/kg, qui correspond à une exposition 3 fois plus importante de BOTOX que celle attendue avec la dose clinique recommandée de 200 unités dans le traitement de l'incontinence urinaire causée par une hyperactivité neurogène du détrusor (basée sur une personne de 50 kg).
Mutagénicité
BOTOX s'est révélé négatif lors de tests de mutation bactérienne inverse, de tests du micro-nucleus sur des cultures de cellules de mammifères et des analyses chromosomiques. BOTOX s'est révélé négatif lors d'un test de micro-nucleus in vivo chez des rongeurs.
Cancérogénicité
Aucune étude de cancérogénicité n'a été effectuée avec BOTOX.
Toxicité sur la reproduction
Dans les études de fertilité, la NOEL après injection intramusculaire de BOTOX était de 4 unités/kg chez les rats mâles et de 8 unités/kg chez les rats femelles et est équivalente à la dose maximale recommandée de 400 unités sur la base du poids corporel (kg). Des doses plus élevées étaient associées à des diminutions de la fertilité liées à la dose. Lorsque la fécondation a eu lieu, il n'y a pas eu d'effets indésirables sur le nombre ou la viabilité des embryons conçus ou conçus à partir de rats mâles ou femelles traités.
Des souris, des rates et des lapines gravides ont reçu pendant la période d'organogenèse des injections de BOTOX par voie intramusculaire. La dose sans effet toxique observé (NOAEL) au cours de la phase de développement était de 4, de 1 et de 0,125 unités/kg de poids corporel, respectivement. Les doses plus élevées ont entraîné une réduction du poids du fœtus et/ou un retard d'ossification. On a constaté des avortements chez le lapin.
Plus de données
Animaux juvéniles
Dans une étude où des rats juvéniles ont reçu une injection intramusculaire de BOTOX à la dose de 0, 8, 16 ou 24 unités/kg de poids corporel toutes les deux semaines à partir du 21e jour postnatal pendant 3 mois, on a observé des changements dans la taille/géométrie des os associés à une diminution de la densité et de la masse osseuse secondaire, à une lésion des membres, à un manque de contraction musculaire et à une diminution du gain de poids corporel. Les changements étaient moins graves à la plus faible dose testée et présentaient des signes de réversibilité à toutes les doses. Les animaux juvéniles semblent plus sensibles que les adultes. Dans une étude où des rats juvéniles ont reçu une injection intramusculaire de BOTOX toutes les deux semaines pendant 12 semaines, la valeur NOAEL(No Observed Adverse Effect Level) était de 8 unités/kg, ce qui est comparable à la dose maximale proposée pour les patients pédiatriques de 10 unités/kg. La dose cumulée chez les rats juvéniles de 56 unités/kg (8 unités/kg administrées toutes les deux semaines pendant 12 semaines) est 5,6 fois supérieure à la dose cumulée maximale dans la population spastique pédiatrique sur la même période. La dose non efficace pour les répercussions négatives sur le développement chez les jeunes animaux (8 unités/kg de poids corporel) correspond à la dose maximale cumulée chez les adultes (400 unités) et est inférieure à la dose maximale pédiatrique (340 unités) corporel sur la base du poids corporel (kg).
Remarques particulièresIncompatibilités
En l'absence d'étude d'incompatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Tenir hors de la portée des enfants.
Flacons fermés: Conserver au réfrigérateur entre 2-8°C ou au congélateur à -5°C ou en dessous.
Les études sur l'activité ont montré que le médicament peut être conservé après reconstitution jusqu'à 3 jours à 2–8°C. Comme le médicament ne contient pas de conservateur, il faut, pour des raisons microbiologiques, utiliser la préparation prête à l'emploi immédiatement après dilution, sauf si cette dernière a été effectuée dans des conditions aseptiques contrôlées et validées. Le médicament ne devrait normalement pas être conservé plus de 24 heures à 2–8°C.
Remarques concernant la manipulation
Préparation de la solution injectable de BOTOX
Il est recommandé d'effectuer la reconstitution de la solution et la préparation de la seringue sur des serviettes en papier doublées de plastique afin d'absorber toute quantité renversée. Reconstituer BOTOX avec une solution saline normale sans conservateur (chlorure de sodium à 0,9% pour préparations injectables).
Prélever une quantité appropriée de diluant (voir table de dilution ci-dessous) dans une seringue. Il convient de nettoyer la surface du bouchon en caoutchouc du flacon avec un tampon imbibé d'alcool avant d'introduire l'aiguille.
Tableau 9: Tableau de dilution pour les flacons contenant BOTOX 50 et 100 unités pour toutes les indications (sauf injections dans le muscle détrusor)
|
|
Flacon de 50 unités
|
Flacon de 100 unités
| |
Dose résultante
(unités par 0,1 ml)
|
Quantité de solvant (chlorure de sodium 9 mg/ml (0,9%)), à ajouter à un flacon de 50 unités:
|
Quantité de solvant (chlorure de sodium 9 mg/ml (0,9%)), à ajouter à un flacon de 100 unités:
| |
20,0 E
|
0,25 ml
|
0,5 ml
| |
10,0 E
|
0,5 ml
|
1,0 ml
| |
5,0 E
|
1 ml
|
2,0 ml
| |
2,5 E
|
2 ml
|
4,0 ml
| |
1,25 E
|
4 ml
|
8,0 ml
|
Vessie hyperactive
Instruction pour la dilution de deux flacons BOTOX de 50 unités:
·Reconstituer deux flacons de BOTOX de 50 unités avec 5 ml chacun de solution de chlorure de sodium à 0,9% (sans agent conservateur) et mélanger le contenu du flacon avec précautions.
·Aspirer 5 ml de chaque flacon dans une seringue de 10 ml.
Il en résulte une seringue de 10 ml contenant au total 100 unités de BOTOX reconstitué. Utiliser immédiatement après reconstitution dans la seringue. Jeter toute solution de chlorure de sodium non utilisée.
Instructions pour la dilution d'un flacon BOTOX de 100 unités:
·Reconstituer un flacon BOTOX de 100 unités avec 10 ml d'une solution de chlorure de sodium à 0,9% (sans agent conservateur) et mélanger le contenu du flacon avec précautions.
·Aspirer 10 ml du flacon dans une seringue de 10 ml.
Il en résulte une seringue de 10 ml contenant au total 100 unités de BOTOX reconstitué. Utiliser immédiatement après reconstitution dans la seringue. Jeter toute solution de chlorure de sodium non utilisée.
Incontinence urinaire causée par une hyperactivité neurogène du détrusor
Instructions pour la dilution de quatre flacons de 50 unités de BOTOX:
·Reconstituer quatre flacons de 50 unités de BOTOX avec 3 ml chacun d'une solution de chlorure de sodium à 0,9% (sans agent conservateur) et mélanger le contenu du flacon avec précautions.
·Aspirer 3 ml du premier flacon et 1 ml du deuxième flacon dans une seringue de 10 ml.
·Aspirer 3 ml du troisième flacon et 1 ml du quatrième flacon dans une deuxième seringue de 10 ml.
·Aspirer les 2 ml restants des deuxième et quatrième flacons dans une troisième seringue de 10 ml.
·Terminer la reconstitution en ajoutant 6 ml de solution saline à 0,9% sans conservateur dans chacune des trois seringues de 10 ml et agiter-les doucement.
Il en résulte trois seringues de 10 ml contenant au total 200 unités de BOTOX reconstitué. Utiliser immédiatement après reconstitution dans la seringue. Jeter toute solution de chlorure de sodium non utilisée.
Instructions pour la dilution de deux flacons de 100 unités de BOTOX:
·Reconstituer deux flacons de 100 unités de BOTOX, chacun à l'aide de 6 ml d'une solution de chlorure de sodium à 0,9% (sans agent conservateur) et mélanger le contenu du flacon avec précautions.
·Aspirer 4 ml du contenu de chacun des flacons dans deux seringues de 10 ml chacune.
·Aspirer les 2 ml restants du contenu de chaque flacon dans une troisième seringue de 10 ml.
·Terminer la reconstitution en ajoutant 6 ml de solution saline à 0,9% sans conservateur dans chacune des seringues de 10 ml et agitez-les doucement.
Il en résulte trois seringues de 10 ml contenant au total 200 unités de BOTOX reconstitué. Utiliser immédiatement après reconstitution dans la seringue. Jeter toute solution de chlorure de sodium non utilisée.
Il est possible d'obtenir une concentration de BOTOX plus ou moins forte en ajoutant un volume de diluant plus ou moins important.
BOTOX étant susceptible d'être dénaturé par la formation de bulles ou une agitation vigoureuse, le diluant doit être injecté délicatement dans le flacon. Ne pas utiliser le flacon si un léger vide n'entraîne pas l'aspiration du diluant à l'intérieur du flacon. Une fois reconstitué, BOTOX est une solution incolore ou légèrement jaune, exempte de particules en suspension.
Utiliser immédiatement après reconstitution dans la seringue. Après reconstitution, il convient d'utiliser la seringue immédiatement. Éliminer la solution de chlorure de sodium non utilisée.
La solution reconstituée est à usage unique. La solution reconstituée de BOTOX, qui est de nouveau diluée dans une seringue pour injection dans le muscle détrusor, doit être utilisée immédiatement. Toute solution non utilisée doit être éliminée de manière appropriée.
Élimination
Si la poudre BOTOX s'est répandue, elle doit être essuyée avec un linge absorbant imbibé de solution diluée d'hypochlorite (à 0,5%). Si la solution injectable BOTOX s'est répandue, elle doit être essuyée avec un linge absorbant sec. Les surfaces souillées doivent être nettoyées avec un linge absorbant imbibé de solution diluée d'hypochlorite, puis séchées en frottant.
Si un flacon-ampoule s'est brisé, les éclats de verre doivent être recueillis prudemment de sorte à éviter toute blessure de la peau.
Des flacons pour injection non utilisés doivent être préparés avec un peu d'eau et autoclavés. Tous les flacons pour injection utilisés, seringues, aiguilles, etc. doivent également être autoclavés. Les résidus de toxine botulique peuvent également être inactivés avec une solution d'hypochlorite diluée à 0,5%. La solution doit être appliquée pendant au moins 5 minutes. Les autres éléments contaminés, p.ex. éclats de verre, gants, etc., doivent être jetés dans un contenant pour déchets tranchants et éliminés par incinération.
Identification du médicament
Si le produit BOTOX utilisé est authentique, des lignes horizontales aux couleurs de l’arc-en-ciel et le nom «abbvie» sont visibles sur l’étiquette (hologramme) en tournant le flacon à droite et à gauche sous une lampe de bureau ou un néon. Si cela n’est pas le cas et/ou si la perforation n’est pas intacte, alors le produit ne doit pas être utilisé et AbbVie doit être contacté.
En outre, l’autocollant avec le numéro de lot et la date d’expiration peut être détaché de l’étiquette du flacon de BOTOX et collé dans le dossier du patient pour faciliter la traçabilité du lot. Après le décollement de l’autocollant de l’étiquette du flacon, la mention «Verwendet/Utilisé» apparaît.
Numéro d’autorisation52433 (Swissmedic)
PrésentationBOTOX
Flacon contenant 50 unités (bague rouge en aluminium): 2 x 1 [A]
Flacon contenant 100 unités (bague violette en aluminium): 1 et 2 x 1 [A]
Titulaire de l’autorisationAbbVie AG, 6330 Cham
Mise à jour de l’informationMai 2025
|