CompositionPrincipes actifs
Sécukinumab*.
*produit à partir de cellules CHO (Chinese Hamster Ovary) génétiquement modifiées
Excipients
Poudre pour solution injectable
Saccharose, L-histidine, chlorhydrate de L-histidine monohydraté, polysorbate 80, pro vitro.
Solution injectable en seringue préremplie ou en stylo prérempli
Tréhalose dihydraté, L-histidine, chlorhydrate de L-histidine monohydraté, L-méthionine, polysorbate 80, eau pour préparation injectable q.s. ad solutionem pro 0,5 ml ou 1 ml ou 2 ml.
Forme pharmaceutique et quantité de principe actif par unitéPour administration sous-cutanée.
Poudre pour solution injectable
Chaque flacon contient 150 mg de sécukinumab après reconstitution avec 1 ml d'eau pour préparation injectable.
Solution injectable en seringue préremplie ou en stylo prérempli
Chaque seringue préremplie de 2 ml ou chaque stylo prérempli de 2 ml contient 300 mg de sécukinumab.
Chaque seringue préremplie de 1 ml ou chaque stylo prérempli de 1 ml contient 150 mg de sécukinumab.
Chaque seringue préremplie de 0,5 ml contient 75 mg de sécukinumab.
Indications/Possibilités d’emploiPsoriasis en plaques
Cosentyx/- SensoReady/- UnoReady est indiqué dans le traitement des patients adultes et des patients pédiatriques âgés de 6 ans et plus atteints de psoriasis en plaques modéré à sévère, qui n'ont pas répondu à d'autres traitements systémiques, notamment la ciclosporine, le méthotrexate ou la PUVA, chez qui ces traitements sont contre-indiqués ou qui ne tolèrent pas ces traitements.
Rhumatisme psoriasique
Cosentyx/- SensoReady/- UnoReady, seul ou en association avec le méthotrexate, est indiqué dans le traitement des patients adultes atteints de rhumatisme psoriasique actif, qui n'ont pas répondu de manière adéquate à un traitement antérieur par des médicaments antirhumatismaux modificateurs de la maladie (disease-modifying anti-rheumatic drugs, DMARD). Cosentyx/- SensoReady/- UnoReady retarde la progression des atteintes structurales et améliore la fonction physique.
Spondylarthrite axiale (axSpA)
Spondylarthrite ankylosante (SA, maladie de Bechterew)
Cosentyx/- SensoReady est indiqué dans le traitement des patients adultes atteints de spondylarthrite ankylosante sévère et active, qui n'ont pas répondu de manière adéquate à un traitement conventionnel (par exemple les AINS).
Spondylarthrite axiale non radiographique (nr-axSpA)
Cosentyx/- SensoReady est indiqué dans le traitement de la spondylarthrite axiale non radiographique sévère et active, avec signes objectifs d'inflammation avec une augmentation de la protéine C-réactive (CRP) et des résultats d'imagerie par résonance magnétique (IRM) chez les adultes qui n'ont pas répondu de manière adéquate à un traitement par des anti-inflammatoires non stéroïdiens (AINS).
Arthrite juvénile idiopathique (AJI)
Arthrite liée à l'enthésite (ERA)
Cosentyx/- SensoReady est indiqué dans le traitement de l'arthrite liée à l'enthésite active chez les patients âgés de 6 ans et plus dont l'affection n'a pas répondu de manière adéquate aux médicaments anti-inflammatoires non stéroïdiens (AINS) et aux antirhumatismaux modificateurs de la maladie (DMARD). Voir "Posologie/Mode d'emploi" et "Propriétés/Effets" .
Rhumatisme psoriasique juvénile (RPJ)
Cosentyx/- SensoReady est indiqué dans le traitement du rhumatisme psoriasique juvénile actif chez les patients âgés de 6 ans et plus dont l'affection a répondu de manière insatisfaisante aux médicaments anti-inflammatoires non stéroïdiens (AINS) et aux antirhumatismaux modificateurs de la maladie (DMARD). Voir "Posologie/Mode d'emploi" et "Propriétés/Effets" .
Hidrosadénite suppurée (HS)
Cosentyx est indiqué dans le traitement des patients adultes atteints d'hidrosadénite suppurée (maladie de Verneuil) active modérée à sévère, qui n'ont pas répondu de manière adéquate à une antibiothérapie systémique.
Posologie/Mode d’emploiL'utilisation de Cosentyx/- SensoReady/- UnoReady doit se faire sous la direction et la supervision d'un médecin expérimenté dans le diagnostic et le traitement des maladies pour lesquelles Cosentyx/ Cosentyx SensoReady/- UnoReady est indiqué.
Cosentyx est administré par injection sous-cutanée (pour la biodisponibilité, voir la rubrique "Pharmacocinétique" ). Les zones de peau présentant des signes de psoriasis doivent être évitées comme sites d'injection.
Après une formation adéquate à la technique d'auto-injection sous-cutanée, les patients peuvent s'auto-injecter Cosentyx/- SensoReady/- UnoReady ou se le faire injecter par un aidant si un médecin considère cela comme approprié. Le médecin doit toutefois assurer un suivi adéquat des patients. Il convient de demander aux patients et/ou aux aidants d'injecter la quantité complète de Cosentyx/- SensoReady/- UnoReady conformément aux instructions figurant dans la notice.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Posologie usuelle
Patients adultes
Psoriasis en plaques
La dose recommandée est de 300 mg en injection sous-cutanée avec des doses initiales aux semaines 0, 1, 2, 3 et 4, suivies de doses d'entretien mensuelles. Chaque dose de 300 mg est administrée sous forme d'une injection sous-cutanée de 300 mg ou de deux injections sous-cutanées de 150 mg.
Certains patients dont le poids corporel est ≥90 kg peuvent tirer un bénéfice supplémentaire de l'administration de 300 mg toutes les 2 semaines.
Rhumatisme psoriasique
La dose recommandée est de 150 mg en injection sous-cutanée avec des doses initiales aux semaines 0, 1, 2, 3 et 4, suivies de doses d'entretien mensuelles. La dose peut être augmentée jusqu'à 300 mg, en fonction de la réponse clinique.
Chez les patients qui répondent de manière inadéquate aux anti-TNF-alpha (répondeurs inadéquats, RI), la dose recommandée est de 300 mg en injection sous-cutanée avec des doses initiales aux semaines 0, 1, 2, 3 et 4, suivies de doses d'entretien mensuelles. Chaque dose de 300 mg est administrée sous forme d'une injection sous-cutanée de 300 mg ou de deux injections sous-cutanées de 150 mg.
Pour les patients présentant un psoriasis en plaques concomitant modéré à sévère, voir les recommandations posologiques et d'utilisation pour le psoriasis en plaques.
Spondylarthrite axiale (axSpA)
Spondylarthrite ankylosante (SA, maladie de Bechterew)
La dose recommandée est de 150 mg en injection sous-cutanée avec des doses initiales aux semaines 0, 1, 2, 3 et 4, suivies de doses d'entretien mensuelles.
Spondylarthrite axiale non radiographique (nr-axSpA)
La dose recommandée est de 150 mg en injection sous-cutanée avec des doses initiales aux semaines 0, 1, 2, 3 et 4, suivies de doses d'entretien mensuelles.
Les données disponibles sur le rhumatisme psoriasique et la spondylarthrite axiale indiquent qu'une réponse clinique est généralement obtenue dans un délai de 16 semaines de traitement. Chez les patients qui n'ont pas répondu au traitement après 16 semaines de traitement, il faut envisager d'arrêter le traitement. Chez certains patients présentant initialement une réponse incomplète, des améliorations ultérieures peuvent être observées si le traitement est poursuivi au-delà de 16 semaines.
Hidrosadénite suppurée
La dose recommandée est de 300 mg de sécukinumab en injection sous-cutanée, dans un premier temps avec des doses initiales aux semaines 0, 1, 2, 3 et 4, suivies de doses d'entretien mensuelles. En cas d'absence de réponse clinique (HiSCR50) après 32 semaines, la dose d'entretien peut être augmentée à 300 mg toutes les 2 semaines. Si aucune réponse clinique n'est obtenue dans les 16 semaines, le traitement doit être arrêté. Chaque dose de 300 mg est administrée soit sous la forme d'une injection sous-cutanée de 300 mg, soit sous la forme de deux injections sous-cutanées de 150 mg chacune.
Patients pédiatriques
Psoriasis en plaques
La dose recommandée dépend du poids corporel et est administrée en injection sous-cutanée avec des doses initiales aux semaines 0, 1, 2, 3 et 4, suivies de doses d'entretien mensuelles. Chaque dose de 75 mg est administrée sous forme d'une injection sous-cutanée de 75 mg. Chaque dose de 150 mg est administrée sous forme d'une injection sous-cutanée de 150 mg.
Tableau 1: Posologie recommandée pour le psoriasis en plaques chez les enfants et les adolescents
Poids corporel au moment de l'administration Dose recommandée
< 50 kg 75 mg
≥50 kg 150 mg
Arthrite juvénile idiopathique (AJI)
Arthrite liée à l'enthésite (ERA) et rhumatisme psoriasique juvénile (RPJ)
La dose recommandée dépend du poids corporel. Pour les patients dont le poids corporel se situe entre 15 kg et < 50 kg, la dose est de 75 mg. Pour les patients dont le poids corporel est ≥50 kg, la dose est de 150 mg. Cosentyx est administré en injection sous-cutanée avec des doses de départ aux semaines 0, 1, 2, 3 et 4, suivies de doses d'entretien mensuelles (toutes les 4 semaines). Chaque dose de 75 mg est administrée sous forme d'une injection sous-cutanée de 75 mg. Chaque dose de 150 mg est administrée sous forme d'une injection sous-cutanée de 150 mg.
Ajustement de la posologie du fait d'effets indésirables/d'interactions
Psoriasis en plaques
En cas d'effets indésirables graves (voir "Mises en garde et précautions" ), une interruption temporaire du traitement doit être envisagée jusqu'à la disparition de l'effet indésirable. Étant donné que, dans les études cliniques, les infections à candida mucocutanées, globalement rares, sont apparues plus fréquemment sous 300 mg, une réduction de la dose à 150 mg doit être envisagée dans les cas graves.
Patients présentant des troubles de la fonction hépatique / troubles de la fonction rénale
Cosentyx/- SensoReady/- UnoReady n'a pas été spécifiquement étudié chez ces groupes de patients.
Patients âgés
On ne dispose que de données limitées sur l'utilisation de Cosentyx chez les patients de plus de 75 ans atteints de rhumatisme psoriasique et chez les patients de plus de 65 ans atteints de spondylarthrite axiale. On ne dispose que de données très limitées sur l'utilisation de Cosentyx chez les patients de plus de 65 ans atteints d'hidrosadénite suppurée. Cependant, rien n'indique qu'une posologie différente soit nécessaire chez les patients âgés par rapport aux patients adultes plus jeunes.
Enfants et adolescents
La sécurité et l'efficacité chez les enfants de moins de 6 ans atteints d'AJI des sous-types ERA et RPJ n'ont pas été établies.
La sécurité et l'efficacité de Cosentyx n'ont pas été étudiées chez les enfants souffrant de psoriasis en plaques âgés de moins de 6 ans.
La sécurité et l'efficacité pour les patients de moins de 18 ans n'ont pas encore été étudiées dans le traitement de l'hidrosadénite suppurée.
Contre-indicationsRéactions d'hypersensibilité sévères au principe actif ou à l'un des excipients (voir "Composition" , "Mises en garde et précautions" et "Effets indésirables" ).
Infections actives graves (p.ex. tuberculose active, sepsis, infections opportunistes graves).
Mises en garde et précautionsInfections
Cosentyx/- SensoReady/- UnoReady peut augmenter le risque d'infection. Dans les études cliniques, des infections ont été observées chez des patients sous Cosentyx (voir "Effets indésirables" ). La plupart des infections étaient de gravité légère ou modérée. L'expérience chez les patients présentant des infections actives connues par le VIH, le VHB ou le VHC est limitée. Par conséquent, avant d'utiliser Cosentyx/- SensoReady/- UnoReady chez ces patients, il convient d'évaluer le bénéfice du traitement par rapport aux risques potentiels.
Dans les études cliniques, des infections à Candida dose-dépendantes ont été observées, y compris la candidose œsophagienne. Cependant, aucune extension systémique n'a été observée, les infections ont été contrôlées avec un traitement standard et n'ont pas nécessité l'arrêt du traitement. Les données des études cliniques menées jusqu'à 1 an ne montrent pas d'augmentation du risque d'infections opportunistes graves. Les données actuelles ne permettent pas d'évaluer le risque à long terme relatif aux infections opportunistes graves.
L'utilisation de Cosentyx/- SensoReady/- UnoReady chez les patients ayant des antécédents d'infection chronique ou d'infections récidivantes doit être envisagée avec prudence.
Il convient de conseiller aux patients de demander un avis médical en cas d'apparition de signes ou de symptômes évocateurs d'une infection. Si un patient développe une infection grave, il doit être étroitement surveillé; Cosentyx/- SensoReady/- UnoReady ne doit pas être administré tant que l'infection n'a pas disparu.
Évaluation d'un traitement préalable de la tuberculose
Une tuberculose (forme active et/ou réactivation d'une forme latente) peut se développer chez les patients traités par Cosentyx/- SensoReady/- UnoReady. Il convient de soumettre les patients à un dépistage de la tuberculose avant le début du traitement par Cosentyx/- SensoReady/- UnoReady. Cosentyx/- SensoReady/- UnoReady ne doit pas être administré aux patients atteints de tuberculose active. Chez les patients atteints de tuberculose latente, un traitement antituberculeux doit être envisagé avant d'instaurer un traitement par Cosentyx/- SensoReady/- UnoReady. Chez les patients ayant des antécédents de tuberculose latente ou active, chez lesquels un traitement adéquat ne peut pas être confirmé, un traitement antituberculeux doit être envisagé avant l'instauration du traitement par Cosentyx/- SensoReady/- UnoReady. Les patients doivent être étroitement surveillés pendant et après le traitement afin de déceler tout signe et symptôme de tuberculose active.
Réactivation de l'hépatite B
Une réactivation du virus de l'hépatite B peut avoir lieu chez les patients traités par Cosentyx/- SensoReady/- UnoReady. En accord avec les directives cliniques concernant les immunosuppresseurs, les patients doivent être testés afin de dépister une infection par le VHB avant de commencer le traitement par Cosentyx/- SensoReady/- UnoReady. Cosentyx/- SensoReady/- UnoReady ne doit pas être administré aux patients présentant une hépatite B active. Les patients présentant une sérologie positive avérée pour le VHB doivent être surveillés pendant le traitement pour détecter les symptômes cliniques et biologiques d'une réactivation du VHB. En cas de réactivation du VHB pendant le traitement par Cosentyx/- SensoReady/- UnoReady, il convient d'envisager l'arrêt du traitement et de traiter les patients selon les directives cliniques.
Maladies inflammatoires chroniques de l'intestin
Des cas isolés de maladies inflammatoires chroniques de l'intestin, parfois graves, ont été observés dans les études cliniques. Dans la plupart des cas, il s'agissait d'exacerbations d'une maladie de Crohn préexistante. Dans de tels cas, il convient de réévaluer soigneusement le traitement et d'envisager de l'arrêter (voir "Posologie/Mode d'emploi" ). Le sécukinumab n'a pas montré d'efficacité dans une étude de phase 2 chez des patients atteints d'une maladie de Crohn active. Étant donné que les patients atteints de psoriasis présentent intrinsèquement un risque plus élevé de maladies inflammatoires chroniques de l'intestin, les données actuelles ne permettent pas d'établir de manière probante un lien de cause à effet avec Cosentyx/- SensoReady/- UnoReady.
Maladie inflammatoire de l'intestin nouvellement apparue
Des cas de maladies inflammatoires de l'intestin nouvellement apparues ont été rapportés après l'autorisation de mise sur le marché.
Maladies malignes
Dans les études cliniques jusqu'à 1 an, le traitement par Cosentyx/- SensoReady/- UnoReady ne montre pas de risque accru de maladies malignes. Les résultats des études sur la sécurité à long terme ne sont pas encore disponibles.
Les patients atteints de psoriasis ayant reçu auparavant un traitement par UV doivent être examinés avant et pendant le traitement par Cosentyx/- SensoReady/- UnoReady afin de vérifier l'absence de tumeurs cutanées.
Réactions d'hypersensibilité
Dans les études cliniques, de rares cas de réactions anaphylactiques et d'angiœdèmes ont été observés chez des patients recevant Cosentyx. Des cas d'angiœdèmes ont aussi été rapportés après la commercialisation. En cas de survenue d'une réaction anaphylactique ou d'une autre réaction allergique grave, l'administration de Cosentyx/- SensoReady/- UnoReady doit être immédiatement interrompue et des mesures thérapeutiques appropriées doivent être mises en place.
Éruption eczémateuse
La survenue d'éruptions eczémateuses graves, dont des éruptions de type dermatite, des cas d'eczéma dyshidrosique et d'érythrodermie (dermite exfoliative) ont été rapportés après la commercialisation chez des patients recevant Cosentyx. Quelques-uns de ces cas ont nécessité une hospitalisation (voir "Effets indésirables" ). Les éruptions eczémateuses ont commencé entre quelques jours et plusieurs mois après la première utilisation de Cosentyx. Il peut être nécessaire d'interrompre le traitement par Cosentyx pour traiter l'éruption eczémateuse. Chez quelques patients, les éruptions eczémateuses ont pu être traitées avec succès pendant la poursuite du traitement par Cosentyx.
Poussée de psoriasis à l'arrêt du traitement ( "rebond" )
En cas d'arrêt du traitement chez les patients ayant répondu, il faut tenir compte du risque de poussée du psoriasis. Dans une étude d'extension portant sur des patients ayant répondu, une poussée du psoriasis a été observée après arrêt de la dose de 150 mg et après arrêt de la dose de 300 mg: jusqu'à 8 semaines après l'arrêt du traitement, une poussée a été observée chez 4% des patients traités par 300 mg et 4,7% des patients traités par 150 mg. Des poussées sous forme de psoriasis pustuleux ont touché respectivement 1,1% et 0,7% des patients précédemment traités par 300 mg et 150 mg.
Vaccinations
Les données sur la réponse à la vaccination par des vaccins vivants ou inactivés chez les patients traités par Cosentyx/- SensoReady/- UnoReady sont limitées. Les données actuelles ne permettent pas d'évaluer dans quelle mesure Cosentyx/- SensoReady/- UnoReady inhibe la réponse immunitaire aux néo-antigènes et/ou aux antigènes de rappel. L'utilisation de vaccins vivants atténués n'est pas recommandée pendant ou juste avant un traitement par Cosentyx/- SensoReady/- UnoReady. Avant l'introduction du traitement par Cosentyx/- SensoReady/- UnoReady, il est recommandé de mettre à jour le statut vaccinal des patients conformément aux lignes directrices actuelles en matière de vaccination; cela inclut, selon la situation, les vaccinations contre la varicelle/les infections zostériennes.
Association avec d'autres médicaments biologiques
L'administration concomitante de Cosentyx/- SensoReady/- UnoReady avec d'autres médicaments biologiques n'a pas été étudiée et n'est pas recommandée.
Personnes sensibles au latex - concerne uniquement les seringues/stylos prérempli(e)s
Le capuchon de l'aiguille de la seringue préremplie peut contenir du caoutchouc sec (latex). La sécurité de l'utilisation de la seringue/du stylo prérempli(e) chez les patients présentant une hypersensibilité au latex n'est pas documentée.
InteractionsInteractions avec des vaccins, voir "Mises en garde et précautions" .
Dans une étude menée chez des patients adultes atteints de psoriasis en plaques, aucune interaction n'a été observée entre le sécukinumab et le midazolam (substrat du CYP3A4). Dans les études portant sur le rhumatisme psoriasique et la spondylarthrite axiale, Cosentyx/- SensoReady/- UnoReady a été administré en même temps que le méthotrexate et/ou des corticostéroïdes; aucune interaction n'a été observée.
L'expression des enzymes hépatiques CYP450 est réprimée par les cytokines qui stimulent l'inflammation chronique. Par conséquent, l'expression du CYP450 pourrait être modifiée si une inhibition des cytokines est initiée avec le sécukinumab.
Effet de Cosentyx/- SensoReady/- UnoReady sur d'autres médicaments
Les patients prenant des médicaments dont la dose est ajustée individuellement et qui sont métabolisés par les CYP450 3A4, 1A2 ou 2C9 (p.ex. atorvastatine, bloqueurs des canaux calciques, théophylline, acénocoumarol, phénprocoumone, phénytoïne, ciclosporine ou benzodiazépines) doivent être contrôlés au début et à la fin du traitement par le sécukinumab et la dose de ces substances doit être ajustée si nécessaire. Compte tenu de sa longue demi-vie d'élimination, l'effet du sécukinumab sur l'activité des enzymes CYP450 peut persister pendant plusieurs semaines après l'arrêt du traitement.
Grossesse, allaitementGrossesse
Il n'existe pas de données suffisantes relatives à l'utilisation de Cosentyx/- SensoReady/- UnoReady pendant la grossesse. Il est connu que les IgG1 humaines traversent la barrière placentaire et le sécukinumab est un anticorps monoclonal IgG1. Il est donc possible que le sécukinumab passe de la mère au fœtus. Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par Cosentyx/- SensoReady/- UnoReady. Les patientes doivent être invitées à utiliser des méthodes de contraception efficaces pendant au moins 20 semaines après le dernier traitement par Cosentyx/- SensoReady/- UnoReady. Les études animales ne montrent pas d'effets nocifs directs ou indirects sur la grossesse, le développement embryonnaire, le développement du fœtus, la naissance ou le développement postnatal (voir "Données précliniques" ). Étant donné que les études de reproduction chez l'animal ne permettent pas toujours de prédire la réaction chez l'homme, Cosentyx/- SensoReady/- UnoReady ne doit être utilisé pendant la grossesse que si le bénéfice l'emporte clairement sur les risques potentiels.
Allaitement
On ne sait pas si le sécukinumab passe dans le lait maternel humain. Comme de nombreux médicaments, dont les anticorps, sont excrétés dans le lait maternel, un risque pour le nouveau-né/jeune enfant ne peut pas être exclu. En raison des dommages possibles pour le nourrisson allaité, il est recommandé de ne pas allaiter pendant le traitement par Cosentyx/- SensoReady/- UnoReady et pendant au moins 20 semaines après la dernière dose. Par conséquent, la décision soit d'arrêter l'allaitement, soit d'arrêter Cosentyx/- SensoReady/- UnoReady chez la mère doit être prise en tenant compte des avantages de l'allaitement pour l'enfant et des avantages du traitement pour la mère.
Fertilité
L'effet de Cosentyx sur la fertilité humaine n'a pas été étudié. Des études menées sur les animaux ne montrent aucun effet néfaste direct ou indirect concernant la fertilité (voir "Données précliniques" ).
Effet sur l’aptitude à la conduite et l’utilisation de machinesL'effet de Cosentyx/- SensoReady/- UnoReady sur l'aptitude à la conduite ou l'utilisation de machines n'a pas été étudié.
Effets indésirablesRésumé du profil de sécurité
Au total, plus de 20 000 patients atteints de différentes indications (psoriasis en plaques, rhumatisme psoriasique, spondylarthrite axiale, hidrosadénite suppurée et autres maladies auto-immunes) ont été traités par Cosentyx dans le cadre d'études cliniques en aveugle et en ouvert, ce qui correspond à une exposition de 34 908 patients-années. Parmi ces patients, 14 000 ont été exposés à Cosentyx pendant au moins un an.
Quatre études de phase III contrôlées contre placebo ont été regroupées pour évaluer la sécurité de Cosentyx chez des patients adultes atteints de psoriasis en plaques par rapport à un placebo, pendant une période allant jusqu'à 12 semaines après l'initiation du traitement. Au total, 2076 patients ont été évalués (692 patients ayant reçu des doses de 150 mg, 690 patients des doses de 300 mg et 694 patients ayant reçu un placebo).
L'évaluation de la sécurité de Cosentyx chez les patients atteints de rhumatisme psoriasique repose sur cinq études contrôlées contre placebo portant sur 2754 patients (1871 patients traités par Cosentyx et 883 patients traités par placebo), la phase contrôlée contre placebo ayant duré au maximum 24 semaines, ceci pour une exposition totale des patients dans les études de 4478 patients-années. Le profil de sécurité chez les patients atteints de rhumatisme psoriasique et traités par Cosentyx est le même que celui observé dans le psoriasis.
L'évaluation de la sécurité de Cosentyx chez les patients atteints de spondylarthrite ankylosante repose sur deux études contrôlées contre placebo portant sur 590 patients (394 patients traités par Cosentyx et 196 patients traités par placebo), la phase contrôlée contre placebo ayant duré au maximum 24 semaines, ceci pour une exposition totale des patients dans les études de 755 patients-années (durée moyenne d'exposition des patients qui ont reçu le sécukinumab: 469 jours dans l'étude AS1 et 460 jours dans l'étude AS2).
L'évaluation de la sécurité de Cosentyx chez les patients atteints de spondylarthrite axiale non radiographique (nr-axSpA) repose sur une étude contrôlée contre placebo portant sur 555 patients (369 patients traités par Cosentyx et 186 patients traités par placebo). La période contrôlée contre placebo a duré 52 semaines au maximum et l'exposition totale des patients dans l'étude était de 588 patients-années (durée moyenne d'exposition chez les patients traités par sécukinumab: 395 jours).
Le profil de sécurité chez les patients atteints de spondylarthrite axiale (spondylarthrite ankylosante et spondylarthrite axiale non radiographique) traités par Cosentyx est identique à celui observé dans le psoriasis.
Effets indésirables en cas d'hidrosadénite suppurée
Cosentyx a été étudié dans deux études contrôlées contre placebo chez 1084 patients atteints d'hidrosadénite suppurée (721 patients traités par Cosentyx et 363 patients traités par placebo), avec une exposition dans l'étude de 825 patients-années au total (durée médiane d'exposition chez les patients traités par le sécukinumab: 307 jours). La période contrôlée contre placebo a duré 16 semaines au maximum. Le profil de sécurité observé chez les patients atteints d'hidrosadénite suppurée traités par Cosentyx pendant toute la période de traitement de 52 semaines au maximum correspond au profil de sécurité connu en cas de psoriasis.
Les effets indésirables les plus fréquemment rapportés étaient des infections des voies aériennes supérieures (le plus souvent des rhinopharyngites et des rhinites). La plupart de ces événements étaient de gravité légère ou modérée. Dans les phases contrôlées contre placebo des études pivots de phase III, la proportion de patients ayant arrêté le traitement en raison d'événements indésirables était d'environ 1,5% dans le bras Cosentyx et de 2,4% dans le bras placebo.
Les effets indésirables observés dans les études cliniques sont présentés dans la liste ci-dessous par classes de systèmes d'organes selon la classification MedDRA. Au sein de chaque classe de systèmes d'organes, les effets indésirables sont classés par ordre de fréquence décroissante. Dans chaque groupe de fréquence, les effets indésirables médicamenteux sont indiqués par ordre de sévérité décroissante. Les catégories de fréquence sont définies comme suit: "très fréquent" (≥1/10); "fréquent" (< 1/10, ≥1/100); "occasionnel" (< 1/100, ≥1/1000); "rare" (< 1/1000, ≥1/10 000); "très rare" (< 1/10 000).
Infections et infestations
Très fréquent: infection des voies aériennes supérieures (17,1%)
Fréquent: herpès buccal.
Occasionnel: candidose orale, candidose de l'œsophage, infections des voies aériennes inférieures, pied d'athlète.
Affections hématologiques et du système lymphatique
Occasionnel: neutropénie.
Affections du système nerveux
Fréquent: céphalées.
Affections oculaires
Occasionnel: conjonctivite.
Affections respiratoires, thoraciques et médiastinales
Fréquent: rhinorrhée.
Affections gastro-intestinales
Fréquent: diarrhée, nausées.
Occasionnel: maladies inflammatoires de l'intestin (notamment maladie de Crohn et colite ulcéreuse).
Affections hépatobiliaires
Occasionnels: enzymes hépatiques augmentées, bilirubine augmentée.
Affections de la peau et du tissu sous-cutané
Occasionnel: Urticaire, dermatite (y compris eczéma).
Rare: dermite exfoliative (des cas ont été rapportés chez des patients chez qui un psoriasis avait été diagnostiqué), vascularite d'hypersensibilité, eczéma dyshidrosique.
Affections du système immunitaire
Rare: réactions anaphylactiques.
Troubles généraux et anomalies au site d'administration
Fréquent: fatigue.
Effets indésirables après commercialisation
Les effets indésirables suivants sont survenus après la mise sur le marché de Cosentyx et ont été décrits dans des rapports de cas spontanés et dans la littérature spécialisée. Comme il s'agit de données volontaires provenant d'une population de taille inconnue, il n'est pas possible de donner une estimation fiable de leur fréquence, c'est pourquoi elles sont classées dans la catégorie "fréquence inconnue" . Les effets indésirables sont rangés par classe de systèmes d'organes de la classification MedDRA. Au sein de chaque classe de systèmes d'organes, les effets indésirables sont classés par ordre de sévérité décroissante.
Infections et infestations
Candidoses muqueuse et cutanée.
Affections de la peau et du tissu sous-cutané
Pyoderma gangraenosum, dermite exfoliative généralisée, angiœdème.
Description de certains effets indésirables
Infections
Dans les phases contrôlées contre placebo des études cliniques sur le psoriasis en plaques (dans lesquelles un total de 1382 patients ont été traités par Cosentyx et 694 patients par placebo pendant une durée allant jusqu'à 12 semaines), des infections ont été rapportées chez 28,7% des patients traités par Cosentyx, contre 18,9% des patients traités par placebo. La plupart de ces infections étaient de sévérité légère ou modérée. Des infections à Candida sont apparues chez 1,2% des patients traités par Cosentyx 300 mg, chez 0,4% des patients traités par Cosentyx 150 mg, et chez 0,3% des patients traités par placebo. Des infections graves sont survenues chez 0,14% des patients traités par Cosentyx et chez 0,3% des patients traités par placebo (voir "Mises en garde et précautions" ).
Sur l'ensemble de la phase de traitement (3430 patients au total, dont la plupart ont été traités par Cosentyx pendant une durée allant jusqu'à 52 semaines), des infections ont été rapportées chez 47,5% des patients traités par Cosentyx (0,9 par patient-année de suivi). Des infections graves ont été rapportées chez 1,2% des patients traités par Cosentyx (0,015 par patient-année de suivi).
Les taux d'infection observés dans les études cliniques portant sur le rhumatisme psoriasique, la spondylarthrite axiale (spondylarthrite ankylosante et spondylarthrite axiale non radiographique) étaient similaires à ceux observés dans les études sur le psoriasis.
En raison de la nature des lésions cutanées, les patients atteints d'hidrosadénite suppurée sont plus vulnérables aux infections. Dans la phase contrôlée contre placebo des études cliniques menées chez des patients atteints d'hidrosadénite suppurée (au total, 723 patients ont été traités par le sécukinumab et 363 patients par placebo, chacun pendant une période maximale de 16 semaines), le nombre d'infections était supérieur à celui des études portant sur le psoriasis (30,8% des patients traités par le sécukinumab contre 31,7% des patients traités par placebo). La plupart de ces infections étaient sans gravité, d'intensité légère ou modérée, et n'ont pas nécessité l'arrêt ou une interruption du traitement. Sur l'ensemble de la phase de traitement (au total 1062 patients traités par Cosentyx pendant une durée maximale de 52 semaines), des infections ont été rapportées chez 45,7% des patients traités par Cosentyx (88,6 pour 100 patients-années). Des infections graves ont été rapportées chez 3,2% des patients traités par Cosentyx (4,2 pour 100 patients-années).
Enzymes hépatiques augmentées, bilirubine augmentée
Au cours de la phase contrôlée contre placebo des études cliniques sur le psoriasis en plaques, 14,5% et 5% des patients sous Cosentyx contre 11,6% et 2,5% sous placebo ont développé des augmentations isolées et transitoires des transaminases hépatiques ou de la bilirubine pouvant atteindre 3 fois la limite supérieure de la normale. Les fréquences sous sécukinumab et étanercept étaient comparables.
Neutropénie
Une neutropénie a été observée plus fréquemment avec le sécukinumab qu'avec le placebo. La plupart des cas ont été légers, transitoires et réversibles. Une neutropénie < 1,0–0,5 × 109/l (CTCAE grade 3) a été rapportée chez 18 des 3430 (0,5%) patients sous sécukinumab, sans dépendance à la dose et sans lien temporel avec des infections dans 15 des 18 cas. Aucun cas de neutropénie plus grave n'a été signalé. Dans les 3 autres cas, des infections non graves ont été rapportées, qui ont répondu avec succès au traitement standard et n'ont pas nécessité l'arrêt de Cosentyx.
La fréquence de la neutropénie dans le rhumatisme psoriasique, la spondylarthrite axiale (spondylarthrite ankylosante et spondylarthrite axiale non radiographique) et l'hidrosadénite suppurée a été similaire à celle observée dans le contexte d'un psoriasis.
Réactions d'hypersensibilité
Sur l'ensemble des études cliniques, de l'urticaire, de rares cas de réactions anaphylactiques et des cas d'angiœdèmes ont été observés chez des patients ayant reçu Cosentyx. Des cas d'angiœdèmes ont aussi été rapportés après la commercialisation.
Maladies inflammatoires chroniques de l'intestin
Sur l'ensemble des études cliniques, quelques cas de maladies inflammatoires chroniques de l'intestin ont été observés sous sécukinumab chez des patients à risque (voir "Mises en garde et précautions" ). Un lien de causalité avec le sécukinumab ne peut pas être exclu sur la base des données actuelles.
Immunogénicité
Dans les études cliniques sur le psoriasis, le rhumatisme psoriasique et la spondylarthrite axiale (spondylarthrite ankylosante et spondylarthrite axiale non radiographique) ainsi que l'hidrosadénite suppurée, moins de 1% des patients traités par Cosentyx ont développé des anticorps contre le sécukinumab pendant un traitement allant jusqu'à 52 semaines. Environ la moitié des anticorps dirigés contre le médicament apparus pendant le traitement étaient des anticorps neutralisants, mais cela n'a pas été associé à une perte d'efficacité ni à des variations des paramètres pharmacocinétiques.
Population pédiatrique
Psoriasis en plaques
La sécurité de Cosentyx a été évaluée dans deux études de phase III chez des patients pédiatriques atteints de psoriasis en plaques. La première étude était une étude en double aveugle, contrôlée contre placebo, portant sur 162 patients âgés de 6 à < 18 ans atteints de psoriasis en plaques sévère. La deuxième étude était une étude en ouvert, portant sur 84 patients âgés de 6 à < 18 ans atteints de psoriasis en plaques modéré à sévère. Le profil de sécurité issu de ces deux études portant sur un total de 287,4 patients-années correspondait au profil de sécurité rapporté chez les patients adultes atteints de psoriasis en plaques.
Arthrite juvénile idiopathique (AJI)
Arthrite liée à l'enthésite (ERA) et rhumatisme psoriasique juvénile (RPJ)
La sécurité de Cosentyx a également été examinée dans une étude de phase III menée chez 86 patients pédiatriques âgés de 2 ans à moins de 18 ans, atteints d'AJI des sous-types ERA et RPJ. Le profil de sécurité de cette étude concordait avec le profil de sécurité des patients adultes.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucun cas de surdosage n'a été rapporté dans les études cliniques.
Dans les études cliniques, des doses allant jusqu'à 30 mg/kg (soit environ 2000 à 3000 mg) ont été administrées par voie intraveineuse sans qu'une toxicité limitant la dose ne se produise.
Traitement
En cas de surdosage, il est recommandé de surveiller le patient pour déceler tout signe ou symptôme d'effets indésirables chez le patient et d'instaurer immédiatement un traitement symptomatique approprié.
Propriétés/EffetsCode ATC
L04AC10
Mécanisme d'action
Le sécukinumab est un anticorps IgG1 entièrement humain qui se lie de manière sélective à la cytokine pro-inflammatoire interleukine 17A (IL-17A) et la neutralise. Le sécukinumab agit de manière ciblée sur l'IL-17A et inhibe son interaction avec le récepteur de l'IL-17, qui est exprimé sur différents types de cellules, notamment les kératinocytes. En conséquence, le sécukinumab inhibe la libération de cytokines pro-inflammatoires, de chimiokines et de médiateurs de lésions tissulaires et réduit les effets induits par l'IL-17A dans les maladies auto-immunes et inflammatoires. Des taux de sécukinumab cliniquement pertinents atteignent la peau et réduisent les marqueurs inflammatoires locaux. Comme conséquence directe, le traitement par le sécukinumab réduit l'érythème, le durcissement (induration) et la desquamation dans les lésions du psoriasis en plaques. Chez les patients atteints de psoriasis en plaques, la production d'IL-17A est fortement régulée à la hausse dans les zones cutanées touchées par les lésions par rapport aux zones cutanées sans lésion. En outre, des cellules produisant de l'IL-17 ont été observées plus fréquemment dans le liquide articulaire des patients atteints de rhumatisme psoriasique. De plus, chez les patients atteints de spondylarthrite axiale, les cellules produisant de l'IL-17 étaient également nettement plus nombreuses dans la moelle osseuse sous-chondrale des facettes articulaires. L'IL-17A est nettement régulée à la hausse dans les lésions associées à l'hidrosadénite suppurée en comparaison avec les patients atteints de psoriasis et les personnes de contrôle saines, et des taux sériques d'IL-17A significativement plus élevés ont été observés chez les patients concernés. Une augmentation du nombre de lymphocytes produisant de l'IL-17A a également été constatée chez des patients atteints de spondylarthrite axiale non radiographique.
L'IL-17A favorise également la réaction inflammatoire dans les tissus, l'infiltration par les neutrophiles, la destruction des os et des tissus ainsi que le remodelage des tissus, notamment l'angiogenèse et la fibrose.
Pharmacodynamique
Dans une étude sur le sécukinumab, les taux de neutrophiles infiltrants et de divers marqueurs associés aux neutrophiles, qui sont élevés dans les zones de peau affectées par les lésions chez les patients atteints de psoriasis en plaques, ont été significativement réduits après une à deux semaines de traitement.
Il a été démontré que le sécukinumab réduit (en une à deux semaines de traitement) le taux de protéine C-réactive; la protéine C-réactive est un marqueur de l'inflammation dans le rhumatisme psoriasique et la spondylarthrite axiale.
Efficacité clinique
Psoriasis
Patients adultes
La sécurité et l'efficacité de Cosentyx ont été évaluées dans quatre études de phase III randomisées, en double aveugle, contrôlées contre placebo et d'une durée d'un an menées chez des patients atteints de psoriasis en plaques modéré à sévère qui n'ont pas répondu à la photothérapie ou au traitement systémique ou qui n'ont pas toléré un tel traitement [ERASURE, FIXTURE, FEATURE, JUNCTURE]. L'efficacité et la sécurité de 150 mg et 300 mg de Cosentyx ont été évaluées par rapport à un placebo ou à l'étanercept. De plus, une étude a évalué un schéma de traitement continu par rapport à un schéma avec interruption du traitement à la semaine 12 et reprise du traitement "à la demande" en cas d'aggravation clinique [SCULPTURE]. Dans ces études, chaque dose de 300 mg a été administrée sous la forme de deux injections sous-cutanées de 150 mg.
Afin d'obtenir une évaluation non biaisée de l'efficacité du sécukinumab dans le traitement du psoriasis, l'utilisation concomitante d'un traitement systémique ou topique du psoriasis ou d'une photothérapie n'était pas autorisée pendant les études.
Parmi les 2403 patients inclus dans les études contrôlées contre placebo, 79% n'avaient pas reçu de traitement biologique préalable; 45% présentaient un échec thérapeutique sous un traitement non biologique, 8% un échec thérapeutique sous un traitement biologique, 6% un échec thérapeutique sous un traitement anti-TNF et 2% un échec thérapeutique sous un traitement antip40 (anti-IL-12/IL-23). Les caractéristiques de la maladie à l'inclusion étaient généralement comparables dans tous les groupes de traitement: la valeur médiane du score de l'indice de surface et de sévérité du psoriasis (PASI, Psoriasis-Area-Severity-Index-Score) à l'inclusion était de 19 à 20, le score IGA mod 2011 au moment de l'inclusion était compris entre "modéré" (62%) et "sévère" (38%), la valeur médiane de la surface corporelle (BSA, Body Surface Area) à l'inclusion était ≥27 et la valeur médiane du score de l'indice de qualité de vie en dermatologie (DLQI, Dermatology Life Quality Index) se situait dans la plage allant de 10 à 12. Environ 15 à 25% des patients des études de phase III présentaient un rhumatisme psoriasique (RP) au début de l'étude.
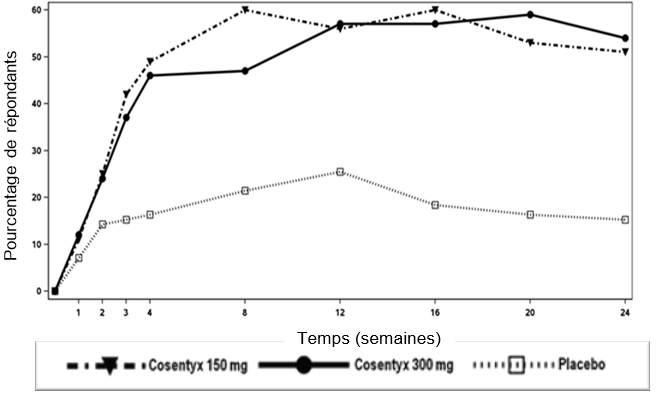
L'étude 1 portant sur le psoriasis (ERASURE) a évalué 738 patients. Les patients randomisés pour recevoir un traitement par Cosentyx ont reçu des doses de 150 mg ou 300 mg aux semaines 0, 1, 2, 3 et 4, suivies de la même dose chaque mois. Les patients randomisés pour recevoir un traitement par placebo et qui étaient non répondeurs à la semaine 12 sont ensuite passés à un traitement par Cosentyx avec des doses de 150 ou 300 mg aux semaines 12, 13, 14 et 15, suivies de la même dose chaque mois à partir de la semaine 16.
L'étude 2 portant sur le psoriasis (FIXTURE) a évalué 1306 patients et a comporté, en plus du groupe placebo, un bras utilisant l'étanercept comme comparateur actif. Le traitement par Cosentyx et placebo était le même que dans l'étude 1. Les patients randomisés pour recevoir un traitement par l'étanercept ont reçu des doses de 50 mg deux fois par semaine pendant 12 semaines, suivies de 50 mg chaque semaine.
Dans l'étude 3 (FEATURE) et l'étude 4 (JUNCTURE) portant sur le psoriasis, 177 patients traités par une seringue préremplie et 182 patients traités par un stylo prérempli ont été comparés à un placebo après 12 semaines de traitement pour évaluer la sécurité, la tolérance et la praticabilité de l'auto-administration de Cosentyx à l'aide de la seringue préremplie. Le traitement par Cosentyx et placebo était le même que dans l'étude 1. Dans l'étude 5 portant sur le psoriasis (SCULPTURE), 966 patients ont été évalués. Tous les patients ont reçu Cosentyx 150 mg ou 300 mg aux semaines 0, 1, 2, 3, 4, 8 et 12, puis ont été randomisés pour suivre soit un traitement d'entretien avec administration continue de la même dose chaque mois, soit, après interruption du traitement, un schéma avec reprise du traitement "à la demande" en cas d'aggravation clinique. Le maintien de la réponse a été moins bon chez les patients ayant interrompu leur traitement et l'ayant repris "à la demande" comparativement aux patients ayant suivi un traitement d'entretien mensuel fixe.
Les critères d'évaluation principaux dans les études contrôlées contre placebo et par agent actif étaient la proportion de patients ayant obtenu une réponse PASI 75 et une réponse "Investigator's Global Assessment (IGA)" mod 2011 dans les catégories "exempt de" ou "presque exempt de" par rapport au placebo à la semaine 12 (voir tableau 2 et tableau 3). Le taux de réponse maximal a été atteint à la semaine 16 et la dose de 300 mg s'est avérée supérieure dans toutes les études.
Tableau 2: Résumé des réponses cliniques PASI 50/75/90/100 & IGA*-mod-2011 dans les catégories "guéri" ou "presque guéri" dans les études sur le psoriasis 1, 3 et 4 (ERASURE, FEATURE et JUNCTURE)
Semaine 12 Semaine 16 Semaine 52
Placebo n (%) (IC à 150 mg n (%) (IC à 300 mg n (%) (IC à 150 mg n (%) (IC à 300 mg n (%) (IC à 150 mg n (%) (IC à 300 mg n (%) (IC à
95%) 95%) 95%) 95%) 95%) 95%) 95%)
Étude 1
Nombre de patients 246 244 245 244 245 244 245
Réponses PASI 50 22 (8,9%) (5,8, 203 (83,5%) (78,1, 222 (90,6%) (86,1, 212 (87,2%) (82,2, 224 (91,4%) (87,0, 187 (77%) (71,0, 207 (84,5%) (79,2,
13,4) 87,9) 93,8) 91,0) 94,5) 82,0) 88,7)
Réponse PASI 75 11 (4,5%) (2,4, 8,1) 174 (71,6%)** 200 (81,6%)** 188 (77,4%) (71,5, 211 (86,1%) (81,0, 146 (60,1%) (53,6, 182 (74,3%) (68,3,
(65,4, 77,1) (76,1, 86,2) 82,4) 90,1) 66,2) 79,5)
Réponse PASI 90 3 (1,2%) (0,3, 3,8) 95 (39,1%)** (33,0, 145 (59,2%)** 130 (53,5%) (47,0, 171 (69,8%) (63,6, 88 (36,2%) (30,2, 147 (60,0%) (53,6,
45,6) (52,7, 65,3) 59,9) 75,4) 42,6) 66,1)
Réponse PASI 100 2 (0,8%) (0,1, 3,2) 31 (12,8%) (9,0, 70 (28,6%) (23,1, 51 (21,0%) (16,2, 102 (41,6%) (35,4, 49 (20,2%) (15,4, 96 (39,2%) (33,1,
17,8) 34,7) 26,8) 48,1) 25,9) 45,6)
Réponse sur l'échell 6 (2,40%) (1,0, 5,5) 125 (51,2%)** 160 (65,3%)** 142 (58,2%) (51,7, 180 (73,5%) (67,4, 101 (41,4%) (35,2, 148 (60,4%) (54,0,
e IGA mod 2011 dans (44,8, 57,6) (58,9, 71,2) 64,4) 78,8) 47,9) 66,5)
les catégories
"exempt de" ou
"presque exempt de"
Étude 3
Nombre de patients 59 59 58 - - - -
Réponse PASI 50 3 (5,1%) (1,3, 15,1) 51 (86,4%) (74,5, 51 (87,9%) (76,1, - - - -
93,6) 94,6)
Réponse PASI 75 0 (0,0%) (0,0, 7,6) 41 (69,5%)** (56,0, 44 (75,9%)** (62,5, - - - -
80,5) 85,7)
Réponse PASI 90 0 (0,0%) (0,0, 7,6) 27 (45,8%) (32,9, 35 (60,3%) (46,6, - - - -
59,2) 72,7)
Réponse PASI 100 0 (0,0%) (0,0, 7,6) 5 (8,5%) (3,2, 19,4) 25 (43,1%) (30,4, - - - -
56,7)
Réponse sur l'échell 0 (0,0%) (0,0, 7,6) 31 (52,5%)** (39,2, 40 (69,0%)** (55,3, - - - -
e IGA mod 2011 dans 65,5) 80,1)
les catégories
"exempt de" ou
"presque exempt de"
Étude 4
Nombre de patients 61 60 60 - - - -
Réponse PASI 50 5 (8,2%) (3,1, 18,8) 48 (80,0%) (67,3, 58 (96,7%) (87,5, - - - -
88,8) 99,4)
Réponse PASI 75 2 (3,3%) (0,6, 12,4) 43 (71,7%)** (58,4, 52 (86,7%)** (74,9, - - - -
82,2) 93,7)
Réponse PASI 90 0 (0,0%) (0,0, 7,4) 24 (40,0%) (27,8, 33 (55,0%) (41,7, - - - -
53,5) 67,7)
Réponse PASI 100 0 (0,0%) (0,0, 7,4) 10 (16,7%) (8,7, 16 (26,7%) (16,5, - - - -
29,0) 39,9)
Réponse sur l'échell 0 (0,0%) (0,0, 7,4) 32 (53,3%)** (40,1, 44 (73,3%)** (60,1, - - - -
e IGA mod 2011 dans 66,1) 83,5)
les catégories
"exempt de" ou
"presque exempt de"
* L'échelle IGA mod
2011 est une échelle
de 5 catégories
avec "0 = exempt
de" , "1 = presque
exempt de" , "2 =
léger" , "3 =
modéré" et "4 =
sévère" et reflète
l'évaluation globale
par le médecin de
la sévérité du
psoriasis en termes
de durcissement,
d'érythème et de
desquamation. Les
réponses "exempt
de" ou "presque
exempt de" , considé
rées comme un
succès thérapeutique
, sont définies
comme l'absence de
signes de psoriasis
ou une coloration
normale à rose des
lésions, l'absence
d'épaississement
des plaques et
l'absence de desquam
ation ou en une
desquamation focale
minimale. ** Valeurs
p versus placebo
et ajustée pour la
multiplicité: p <
0,0001
Tableau 3: Résumé de la réponse clinique dans l'étude 2 portant sur le psoriasis (FIXTURE)
Semaine 12 Semaine 16 Semaine 52
Placebo n (%) (IC à 150 mg n (%) (IC à 300 mg n (%) (IC à Étanercept n (%) 150 mg n (%) (IC à 300 mg n (%) (IC à Étanercept n (%) 150 mg n (%) (IC à 300 mg n (%) (IC à Étanercept n (%)
95%) 95%) 95%) (IC à 95%) 95%) 95%) (IC à 95%) 95%) 95%) (IC à 95%)
Nombre de patients 324 327 323 323 327 323 323 327 323 323
Réponse PASI 50 49 (15,1%) (11,5; 266 (81,3%) (76,6; 296 (91,6%) (87,9; 226 (70,0%) (64,6; 290 (88,7%) (84,6; 302 (93,5%) (90,1; 257 (79,6%) (74,7; 249 (76,1%) (71,1; 274 (84,8%) (80,3; 234 (72,4%) (67,2;
19,6) 85,3) 94,3) 74,9) 91,8) 95,8) 83,7) 80,6) 88,5) 77,2)
Réponse PASI 75 16 (4,9%) (2,9; 8,1) 219 (67,0%)** 249 (77,1%)** 142 (44,0%) (38,5; 247 (75,5%) (70,4; 280 (86,7%) (82,4; 189 (58,5%) (52,9; 215 (65,7%)** 254 (78,6%)** 179 (55,4%) (49,8;
(61,5; 72,0) (72,0; 81,5) 49,6) 80,0) 90,1) 63,9) (60,3; 70,8) (73,7; 82,9) 60,9)
Réponse PASI 90 5 (1,5%) (0,6; 3,8) 137 (41,9%) (36,5; 175 (54,2%) (48,6; 67 (20,7%) (16,5; 176 (53,8%) (48,3; 234 (72,4%) (67,2; 101 (31,3%) (26,3; 147 (45,0%) (39,5; 210 (65,0%) (59,5; 108 (33,4%) (28,4;
47,5) 59,7) 25,7) 59,3) 77,2) 36,7) 50,5) 70,2) 38,9)
Réponse PASI 100 0 (0%) (0,0; 1,5) 47 (14,4%) (10,8; 78 (24,1%) (19,7; 14 (4,3%) (2,5; 7,3) 84 (25,7%) (21,1; 119 (36,8%) (31,6; 24 (7,4%) (4,9; 65 (19,9%) (15,8; 117 (36,2%) (31,0; 32 (9,9%) (7,0;
18,8) 29,3) 30,8) 42,4) 11,0) 24,7) 41,8) 13,8)
Réponse sur l'échell 9 (2,8%) (1,4; 5,4) 167 (51,1%)** 202 (62,5%)** 88 (27,2%) (22,5; 200 (61,2%) (55,6; 244 (75,5%) (70,4; 127 (39,3%) (34,0; 168 (51,4%)** 219 (67,8%)** 120 (37,2%) (31,9;
e IGA mod 2011 dans (45,5; 56,6) (57,0; 67,8) 32,5) 66,4) 80,1) 44,9) (45,8; 56,9) (62,4; 72,8) 42,7)
les catégories
"exempt de" ou
"presque exempt de"
** Valeurs p ajustée
s versus étanercept:
p = 0,0250
Une étude supplémentaire portant sur le psoriasis (CLEAR) a évalué 676 patients. Le sécukinumab 300 mg a atteint les critères d'évaluation principaux et secondaires grâce à sa supériorité sur l'ustékinumab en termes d'ampleur de la réponse PASI 90 à la semaine 16 (critère d'évaluation principal) et de réponse PASI 90 à long terme à la semaine 52. Une efficacité supérieure du sécukinumab par rapport à l'ustékinumab pour les critères d'évaluation réponses PASI 75/90/100 et IGA mod 2011 0 ou 1 ( "exempt de" ou "presque exempt de" ) a été rapidement visible et s'est poursuivie jusqu'à la semaine 52. Dans cette étude, chaque dose de 300 mg a été administrée sous la forme de deux injections sous-cutanées de 150 mg.
Tableau 4: Résumé de la réponse clinique dans l'étude CLEAR
Semaine 16 Semaine 52
Sécukinumab 300 mg Ustékinumab* Sécukinumab 300 mg Ustékinumab*
Nombre de patients 334 335 334 335
Réponse PASI 75 n 311 (93,1%) 276 (82,4%) 306 (91,6%) 262 (78,2%)
(%)
Réponse PASI 90 n 264 (79,0%)** 192 (57,3%) 250 (74,9%)*** 203 (60,6%)
(%)
Réponse PASI 100 n 148 (44,3%) 95 (28,4%) 150 (44,9%) 123 (36,7%)
(%)
Réponse sur l'échell 278 (83,2%) 226 (67,5%) 261 (78,1%) 213 (63,6%)
e IGA mod 2011 dans
les catégories
"exempt de" ou
"presque exempt de"
*Les patients
traités par le
sécukinumab ont
reçu une dose de
300 mg aux semaines
0, 1, 2, 3 et 4,
suivie de la même
dose chaque mois
jusqu'à la semaine
52. Les patients
traités par l'ustéki
numab ont reçu 45
mg ou 90 mg aux
semaines 0 et 4,
puis toutes les 12
semaines jusqu'à la
semaine 52 (dose
déterminée en
fonction du poids
et selon la posologi
e approuvée). **
Valeurs p versus
ustékinumab: p <
0,0001 pour le
critère d'évaluation
principal PASI 90
à la semaine 16 ***
Valeurs p versus
ustékinumab: p =
0,0001 pour le
critère d'évaluation
secondaire PASI 90
à la semaine 52
Cosentyx a été efficace chez les patients non prétraités par un médicament biologique, chez les patients prétraités par un médicament biologique et chez les patients en échec thérapeutique sous un médicament biologique. Les taux de réponse relatifs aux critères d'évaluation principaux, PASI 75 et IGA 0 ou 1, avec Cosentyx 300 mg étaient de 67,7% et 54,1% chez les patients après échec d'un traitement antérieur par inhibiteur du TNF, contre 78,5% et 56,9% chez les patients sans traitement antérieur par inhibiteur du TNF.
Cosentyx, à la dose de 300 mg, a été associé à un début d'action rapide avec une réduction de 50% du PASI moyen à la semaine 3.
Dans toutes les études de phase III portant sur le psoriasis en plaques, environ 15 à 25% des patients inclus présentaient un rhumatisme psoriasique concomitant au début de l'étude. Les améliorations du PASI 75 dans ce groupe de patients ont été comparables à celles observées dans le groupe global de patients atteints de psoriasis en plaques.
Localisation spécifique/formes de psoriasis en plaques
Dans une étude supplémentaire contrôlée contre placebo, une amélioration a été constatée dans le psoriasis unguéal (TRANSFIGURE, 198 patients). Dans l'étude TRANSFIGURE, le sécukinumab a montré un effet supérieur statistiquement significatif par rapport au placebo à la semaine 16 (46,1% pour 300 mg, 38,4% pour 150 mg vs 11,7% pour le placebo) en termes d'amélioration de l'indice de sévérité du psoriasis unguéal (réponse NAPSI, Nail Psoriasis Severity Index) chez des patients atteints de psoriasis en plaques modéré à sévère avec atteinte des ongles.
Dans d'autres études cliniques, des améliorations ont également été observées dans le psoriasis unguéal, l'atteinte du cuir chevelu et l'atteinte palmo-plantaire.
Dans ces études, chaque dose de 300 mg a été administrée sous la forme de deux injections sous-cutanées de 150 mg.
Qualité de vie / résultats rapportés par les patients
Pour le DLQI (Dermatology Life Quality Index), des améliorations statistiquement significatives ont été montrées à la semaine 12 (études 1 à 4) par rapport à la valeur initiale; les améliorations se sont maintenues pendant 52 semaines (études 1 et 2).
Des améliorations statistiquement significatives des signes et symptômes rapportés par les patients - démangeaisons, douleurs et desquamation - à la semaine 12 par rapport à la valeur initiale, en comparaison avec le placebo (études 1 et 2), ont été montrées à l'aide du Psoriasis Symptom Diary© validé.
Dans le DLQI, des améliorations statistiquement significatives par rapport à la valeur initiale ont été observées à la semaine 4 chez les patients traités par le sécukinumab par rapport à ceux traités par l'ustékinumab (CLEAR) et ces améliorations se sont maintenues jusqu'à 52 semaines.
Dans le Psoriasis Symptom Diary, des améliorations statistiquement significatives des signes et symptômes rapportés par les patients - démangeaisons, douleurs et desquamation - ont été observées à la semaine 16 et à la semaine 52 (CLEAR) chez les patients traités par le sécukinumab par rapport à ceux traités par l'ustékinumab.
Patients pédiatriques
Psoriasis en plaques sévère
Une étude de phase III de 52 semaines, randomisée, en double aveugle, contrôlée contre placebo et étanercept a été menée chez 162 patients pédiatriques âgés de 6 à < 18 ans atteints de psoriasis en plaques sévère (défini par un score PASI ≥20, un score IGA mod 2011 de 4 et une surface corporelle affectée ≥10%), pour lesquels un traitement systémique était envisageable. Environ 43% des patients avaient été traités auparavant par photothérapie, 53% par des thérapies systémiques conventionnelles et 3% par des médicaments biologiques. 9% des patients avaient un rhumatisme psoriasique concomitant.
Les patients ont été randomisés pour recevoir l'un des quatre traitements suivants:
-Faible dose de sécukinumab (75 mg pour un poids corporel < 50 kg ou 150 mg pour un poids corporel ≥50 kg) aux semaines 0, 1, 2, 3 et 4, suivie de la même dose toutes les 4 semaines
-Dose élevée de sécukinumab (75 mg pour un poids corporel < 25 kg, 150 mg pour un poids corporel ≥25 kg et < 50 kg ou 300 mg pour un poids corporel ≥50 kg) aux semaines 0, 1, 2, 3 et 4, suivie de la même dose toutes les 4 semaines
-Placebo aux semaines 0, 1, 2, 3 et 4, suivi de la même dose toutes les 4 semaines
-Etanercept (0,8 mg/kg) chaque semaine (jusqu'à un maximum de 50 mg)
Les patients randomisés pour recevoir un placebo et qui n'avaient pas répondu à la semaine 12 ont été placés dans le groupe sécukinumab à faible dose ou à dose élevée (dose basée sur le groupe de poids corporel) et ont reçu le médicament à l'étude aux semaines 12, 13, 14 et 15, puis la même dose toutes les 4 semaines, en commençant à la semaine 16.
Les co-critères d'évaluation principaux étaient la proportion de patients ayant obtenu une amélioration d'au moins 75% du score PASI (réponse PASI 75) et un score IGA mod 2011 "sans symptômes" ou "presque sans symptômes" (0 ou 1) avec une amélioration d'au moins 2 points entre l'inclusion et la semaine 12.
Au cours de la période de 12 semaines contrôlée contre placebo, l'efficacité du sécukinumab à faible dose et à dose élevée était comparable en ce qui concerne les co-critères d'évaluation principaux. Les estimations du rapport des risques en faveur des deux doses de sécukinumab étaient cliniquement pertinentes et statistiquement significatives, tant pour la réponse PASI 75 que pour la réponse IGA mod 2011 de "sans symptôme" ou "presque sans symptôme" (0 ou 1).
Tous les patients ont été suivis pendant 52 semaines après la première dose en ce qui concerne l'efficacité et la sécurité. La proportion de patients ayant obtenu une réponse PASI 75 et une réponse IGA mod 2011 "sans symptôme" ou "presque sans symptôme" (0 ou 1) montrait déjà une différence entre les groupes de traitement par le sécukinumab et le placebo à la semaine 4, la différence s'accentuant à la semaine 12. La réponse s'est maintenue tout au long de la période de 52 semaines. Les scores PASI 50, PASI 90 et PASI 100 et la proportion de patients ayant un score au Children's Dermatology Life Quality Index (CDLQI) de 0 ou 1 se sont également améliorés et ont été maintenus tout au long de la période de 52 semaines.
Après la semaine 12, l'efficacité des doses faibles et élevées de sécukinumab était comparable, bien que l'efficacité de la dose élevée ait été supérieure chez les patients dont le poids corporel était ≥50 kg. Les profils de sécurité des faibles doses et des doses élevées étaient comparables.
Les résultats concernant l'efficacité à la semaine 12 sont présentés dans le tableau 5.
Tableau 5: Résumé de la réponse clinique chez les enfants et adolescents atteints de psoriasis sévère à la semaine 12*
Critère de réponse Comparaison des "Test" "Témoin" Rapport des risques
traitements
"Test" vs "Témoin" n/m** (%) n/m** (%) Évaluateur (IC à valeur p
95%)
à la semaine 12***
PASI 75 Sécukinumab à 32/40 (80,0) 6/41 (14,6) 25,78 (7,08; 114,66) < 0,0001
faible dose vs
placebo
Sécukinumab à dose 31/40 (77,5) 6/41 (14,6) 22,65 (6,31; 98,93) < 0,0001
élevée vs placebo
IGA 0/1 Sécukinumab à 28/40 (70,0) 2/41 (4,9) 51,77 (10,02; < 0,0001
faible dose vs 538,64)
placebo
Sécukinumab à dose 24/40 (60,0) 2/41 (4,9) 32,52 (6,48; 329,52) < 0,0001
élevée vs placebo
PASI 90 Sécukinumab à 29/40 (72,5) 1/41 (2,4) 133,67 (16,83; < 0,0001
faible dose vs 6395,22)
placebo
Sécukinumab à dose 27/40 (67,5) 1/41 (2,4) 102,86 (13,22; < 0,0001
élevée vs placebo 4850,13)
* En cas de données
manquantes, une
imputation en tant
que non répondeur a
été effectuée ** n
= nombre de répondeu
rs, m = nombre de
patients évaluables
*** fenêtre de
temps prolongée
pour les visites à
la semaine 12 Le
rapport des risques,
l'intervalle de
confiance à 95% et
la valeur p provienn
ent d'un modèle de
régression exact
utilisant le groupe
de traitement, la
catégorie de poids
corporel à l'inclusi
on et la catégorie
d'âge comme facteurs
.
Une proportion plus élevée de patients pédiatriques traités par le sécukinumab a signalé une amélioration de la qualité de vie liée à la santé, mesurée par un score CDLQI de 0 ou 1, par rapport au placebo à la semaine 12.
Psoriasis en plaques modéré à sévère
Une étude de phase III multicentrique, en ouvert, à deux bras et en parallèle a été menée chez 84 patients pédiatriques âgés de 6 à < 18 ans atteints de psoriasis en plaques modéré à sévère (défini par un score PASI ≥12, un score IGA mod 2011 ≥3 et une surface corporelle affectée ≥10%) et pour lesquels un traitement systémique était envisageable.
Les patients ont été randomisés pour recevoir un traitement par le sécukinumab aux semaines 0, 1, 2, 3 et 4, suivi de la même dose toutes les 4 semaines, de la manière suivante:
-Faible dose de sécukinumab (75 mg pour un poids corporel < 50 kg ou 150 mg pour un poids corporel ≥50 kg),
-Dose élevée de sécukinumab (75 mg pour un poids corporel < 25 kg, 150 mg pour un poids corporel ≥25 kg et < 50 kg ou 300 mg pour un poids corporel ≥50 kg).
Les co-critères d'évaluation principaux étaient la proportion de patients ayant obtenu une amélioration d'au moins 75% du score PASI (réponse PASI 75) ou un score IGA mod 2011 "sans symptômes" ou "presque sans symptômes" (0 ou 1) avec une amélioration d'au moins 2 points entre l'inclusion et la semaine 12.
L'efficacité du sécukinumab, tant à faible dose qu'à dose élevée, a été comparable et a montré une amélioration cliniquement pertinente pour les co-critères d'évaluation principaux, en comparaison historique par rapport au placebo.
L'efficacité a été évaluée, pour tous les patients, pendant au moins 24 semaines après la première administration. L'efficacité (définie par la réponse PASI 75 et la réponse IGA mod 2011 de "sans symptôme" ou "presque sans symptôme" [0 ou 1]) a été observée dès la semaine 2. La proportion de patients ayant obtenu une réponse PASI 75 et une réponse IGA mod 2011 "sans symptôme" ou "presque sans symptôme" (0 ou 1) a augmenté tout au long de la période de 24 semaines. Des améliorations du PASI 90 et du PASI 100 ont également été observées à la semaine 12 et ont augmenté tout au long de la période de 24 semaines.
Après la semaine 12, l'efficacité des doses faibles et élevées de sécukinumab était comparable. Les profils de sécurité des faibles doses et des doses élevées étaient également comparables.
Les résultats concernant l'efficacité aux semaines 12 et 24 sont présentés dans le tableau 6.
Tableau 6: Résumé de la réponse clinique en cas de psoriasis pédiatrique modéré à sévère aux semaines 12* et 24*
Semaine 12 Semaine 24
Sécukinumab faible Sécukinumab dose Sécukinumab faible Sécukinumab dose
dose élevée dose élevée
Nombre de patients 42 42 42 42
Réponse PASI 75 n 39 (92,9%) 39 (92,9%) 40 (95,2%) 40 (95,2%)
(%)
Réponse IGA mod 33 (78,6%) 35 (83,3%) 37 (88,1%) 39 (92,9%)
2011 "sans symptôme"
ou "presque sans
symptôme" n (%)
Réponse PASI 90 n 29 (69,0%) 32 (76,2%) 37 (88,1%) 37 (88,1%)
(%)
Réponse PASI 100 n 25 (59,5%) 23 (54,8%) 28 (66,7%) 28 (66,7%)
(%)
* En cas de données
manquantes, une
imputation en tant
que non répondeur a
été effectuée.
Flexibilité posologique en cas de psoriasis en plaques
Dans une étude multicentrique, randomisée et en double aveugle menée chez 331 patients, l'efficacité, la sécurité et la tolérance de Cosentyx 300 mg, utilisé toutes les 4 semaines, ont été comparées à celles de Cosentyx 300 mg, utilisé toutes les 2 semaines, chez des patients adultes présentant un poids corporel ≥90 kg et un psoriasis en plaques modéré à sévère. Les patients ont été randomisés selon un rapport 1:1 comme suit:
-Sécukinumab 300 mg aux semaines 0, 1, 2, 3 et 4, suivi de la même dose toutes les 2 semaines jusqu'à la semaine 52 (n = 165).
-Sécukinumab 300 mg aux semaines 0, 1, 2, 3 et 4, suivi de la même dose toutes les 4 semaines jusqu'à la semaine 16 (n = 166).
-Les patients qui avaient été randomisés dans le groupe sécukinumab 300 mg toutes les 4 semaines et avaient atteints une réponse PASI 90 à la semaine 16 ont continué à recevoir le même schéma posologique jusqu'à la semaine 52. Les patients qui avaient été randomisés dans le groupe Cosentyx 300 mg toutes les 4 semaines et qui n'avaient pas atteint une réponse PASI 90 à la semaine 16 ont continué à recevoir le même schéma posologique ou sont passés à Cosentyx 300 mg toutes les 2 semaines pour la période allant jusqu'à la semaine 52.
Les critères d'évaluation principaux et les principaux critères d'évaluation secondaires étaient la proportion de patients qui avaient obtenu, à la semaine 16, une réponse PASI 90 ainsi qu'une réponse allant de "sans symptôme" à "presque sans symptôme" (0 ou 1) dans les catégories IGA mod 2011.
À la semaine 16, la proportion de patients qui avaient obtenu une réponse PASI 90 était plus élevée dans le groupe recevant le traitement toutes les 2 semaines (Q2W) que dans le groupe recevant le traitement toutes les 4 semaines (Q4W) (73,2% et 55,5%, respectivement). La différence entre les traitements était statistiquement significative (valeur p unilatérale = 0,0003).
Le groupe de patients qui n'avaient pas obtenu de réponse PASI 90 et qui étaient passés au schéma de traitement toutes les 2 semaines à la semaine 16 présentaient, à la semaine 32, une réponse PASI 90 plus élevée que dans le groupe de patients qui étaient restés au schéma de traitement toutes les 4 semaines (38,7% et 16,5%, respectivement). La différence entre les traitements était cliniquement pertinente, mais uniquement exploratoire.
Les profils de sécurité des deux schémas posologiques, Cosentyx 300 mg toutes les 4 semaines et Cosentyx 300 mg toutes les 2 semaines, étaient comparables chez les patients présentant un poids corporel ≥90 kg et correspondaient au profil de sécurité rapporté pour les patients atteints de psoriasis.
Rhumatisme psoriasique
Chez les patients adultes atteints de rhumatisme psoriasique actif, il a été démontré que Cosentyx améliore les signes et les symptômes, la fonction physique et la qualité de vie liée à la santé, et réduit en outre le taux de progression des lésions articulaires périphériques.
La sécurité et l'efficacité de Cosentyx ont été démontrées dans trois études de phase III randomisées, en double aveugle et contrôlées contre placebo menées chez 1999 patients qui présentaient un rhumatisme psoriasique actif (≥3 articulations gonflées et ≥3 articulations sensibles à la pression) malgré un traitement par anti-inflammatoires non stéroïdiens (AINS), corticostéroïdes ou médicaments antirhumatismaux modificateurs de la maladie (DMARD). Le diagnostic de RP des patients de ces études avait été posé depuis au moins cinq ans. La majorité des patients présentaient également une lésion de la peau due à un psoriasis actif ou des antécédents documentés de psoriasis. Afin d'obtenir une évaluation non biaisée de l'efficacité de Cosentyx dans le traitement du psoriasis, l'utilisation concomitante d'un traitement topique par corticostéroïdes ou d'une thérapie à base d'UV n'était pas autorisée pendant les études. Plus de 61% et 42% des patients atteints de RP présentaient respectivement une enthésite et une dactylite à l'inclusion. Le nombre de patients atteints de RP avec une atteinte axiale était trop faible pour permettre une évaluation significative.
L'efficacité et la sécurité de Cosentyx aux doses de 75 mg, 150 mg et 300 mg ont été évaluées par rapport à un placebo avec une dose initiale soit par voie intraveineuse soit par voie sous-cutanée. Dans l'étude 1 portant sur le rhumatisme psoriasique (étude PsA1) ou l'étude 2 portant sur le rhumatisme psoriasique (étude PsA2) et l'étude 3 portant sur le rhumatisme psoriasique (étude PsA3), respectivement 29%, 35% et 30% des patients avaient été précédemment traités par des médicaments anti-TNF-alpha, ce traitement ayant été interrompu soit en raison d'une absence de réponse, soit en raison d'une intolérance (patients anti-TNF-alpha-IR).
Dans l'étude PsA1 (FUTURE 1), 606 patients ont été évalués; 60,7% d'entre eux recevaient du MTX de manière concomitante. Des patients présentant tous les sous-groupes de RP ont été inclus, notamment l'arthrite polyarticulaire sans détection de nodules rhumatoïdes (76,7%), la spondylarthrite périphérique (18,5%), l'arthrite périphérique asymétrique (60,2%), l'atteinte interphalangienne distale (59,6%) et l'arthrite mutilante (7,9%). Les patients randomisés pour recevoir Cosentyx ont reçu 10 mg/kg par voie IV aux semaines 0, 2 et 4, suivis d'une dose mensuelle de 75 mg ou 150 mg SC, à partir de la semaine 8. Les patients randomisés pour recevoir un placebo et qui n'ont pas répondu au traitement sont ensuite passés au traitement par 75 mg ou 150 mg de Cosentyx SC une fois par mois à la semaine 16. Au cours de la semaine 24, les patients encore sous placebo ont été transférés vers le traitement par Cosentyx 75 mg ou 150 mg SC. Le critère d'évaluation principal était la réponse selon l'American College of Rheumatology (ACR) 20 à la semaine 24.
L'étude PsA2 (FUTURE 2) a évalué 397 patients, dont 46,6% étaient traités de manière concomitante par MTX. Des patients présentant tous les sous-groupes de RP ont été inclus, notamment l'arthrite polyarticulaire sans détection de nodules rhumatoïdes (85,9%), la spondylarthrite périphérique (21,7%), l'arthrite périphérique asymétrique (64,0%), l'atteinte interphalangienne distale (57,9%) et l'arthrite mutilante (6,3%). Les patients randomisés pour recevoir Cosentyx ont reçu 75 mg, 150 mg ou 300 mg SC aux semaines 0, 1, 2, 3 et 4, puis la même dose chaque mois. Les patients randomisés pour recevoir un placebo et qui n'avaient pas répondu au traitement à la semaine 16 sont ensuite passés à Cosentyx 150 mg ou 300 mg SC une fois par mois à la semaine 16. Au cours de la semaine 24, les patients encore sous placebo ont été transférés vers le traitement par Cosentyx 150 mg ou 300 mg SC. Le critère d'évaluation principal était la réponse ACR 20 à la semaine 24.
L'étude PsA3 (FUTURE 5) a évalué 996 patients, dont 50,1% étaient traités de manière concomitante par MTX. Des patients présentant tous les sous-groupes de RP ont été inclus, notamment l'arthrite polyarticulaire sans détection de nodules rhumatoïdes (78,7%), la spondylarthrite périphérique (19,8%), l'arthrite périphérique asymétrique (65,0%), l'atteinte interphalangienne distale (56,7%) et l'arthrite mutilante (6,8%). Les patients ont été randomisés dans les groupes suivants: Cosentyx 150 mg, Cosentyx 300 mg ou placebo, respectivement par voie SC aux semaines 0, 1, 2, 3 et 4, puis la même dose chaque mois, ou Cosentyx 150 mg une fois par mois sans dose de charge initiale. Les patients initialement randomisés pour recevoir un placebo et n'ayant pas répondu au traitement à la semaine 16 sont ensuite passés à un traitement par Cosentyx (150 mg ou 300 mg SC) une fois par mois à la semaine 16. Au cours de la semaine 24, les patients encore sous placebo ont été transférés vers le traitement par Cosentyx (150 mg ou 300 mg) une fois par mois. Le critère d'évaluation principal était la réponse ACR 20 à la semaine 16 et le principal critère d'évaluation secondaire était la différence du score total de Sharp modifié (mTSS) à la semaine 24 par rapport à l'inclusion.
L'étude PsA4 (FUTURE 3) a évalué 414 patients, dont 47,6% étaient traités de manière concomitante par MTX. Les patients randomisés dans le groupe Cosentyx ont reçu 150 mg ou 300 mg SC aux semaines 0, 1, 2, 3 et 4, puis la même dose chaque mois. Les patients randomisés pour recevoir un placebo et qui n'avaient pas répondu au traitement à la semaine 16 sont ensuite passés à Cosentyx 150 mg ou 300 mg SC une fois par mois à la semaine 16. Au cours de la semaine 24, les patients encore sous placebo ont été transférés vers le traitement par Cosentyx 150 mg ou 300 mg SC. Le critère d'évaluation principal était la réponse ACR 20 à la semaine 24.
Réponse clinique
Signes et symptômes
Le traitement par Cosentyx a entraîné une amélioration significative de l'ampleur de l'activité de la maladie aux semaines 16, 24 et 52 par rapport au placebo. Ces mesures comprenaient la réponse des symptômes articulaires en termes d'ACR 20, ACR 50, ACR 70, la réponse des symptômes cutanés (Psoriasis Area and Severity Index, PASI) 75, PASI 90, ainsi que d'autres scores d'activité de la maladie et d'état de santé (Disease Activity Score, DAS28-CRP, Short Form Health Survey - Physical Component Summary; SF-36 PCS, Health Assessment Questionnaire - Disability Index, HAQ-DI) (voir tableau 7).
Tableau 7: Réponse clinique dans les études PsA2 et PsA3 aux semaines 16, 24 et 52
PsA2 PsA3
Placebo 150 mg1 300 mg1 Placebo 150 mg1 300 mg1
Nombre de patients 98 100 100 332 220 222
randomisés
Réponse ACR 20 n (%)
Semaine 16 18 (18,4%) 60 (60,0%***) 57 (57,0%***) 91◊ (27,4%) 122◊ (55,5%***) 139◊ (62,6%***)
Semaine 24 15◊ (15,3%) 51◊ (51,0%***) 54◊ (54,0%***) 78 (23,5%) 117 (53,2%***) 141 (63,5%***)
Semaine 52 - 64 (64,0%) 64 (64,0%) NA NA NA
Réponse ACR 50 n (%)
Semaine 16 6 (6,1%) 37 (37,0%***) 35 (35,0%***) 27 (8,1%) 79 (35,9%***) 88 (39,6%***)
Semaine 52 - 39 (39,0%) 44 (44,0%) NA NA NA
Réponse ACR 70 n (%)
Semaine 16 2 (2,0%) 17 (17,0%**) 15 (15,0%**) 14 (4,2%) 40 (18,2%***) 45 (20,3%***)
Semaine 52 - 20 (20,0%) 24 (24,0%) NA NA NA
DAS28-CRP
Semaine 16 -0,50 -1,45*** -1,51*** -0,63 -1,29*** -1,49***
Semaine 52 - -1,69 -1,78 NA NA NA
Réponse PASI 75 n
(%)
Semaine 16 3 (7,0%) 33 (56,9%***) 27 (65,9%***) 20 (12,3%) 75 (60,0%***) 77 (70,0%***)
Semaine 52 - 33 (56,9%) 30 (73,2%) - - -
Réponse PASI 90 n
(%)
Semaine 16 3 (7,0%) 22 (37,9%***) 18 (43,9%***) 15 (9,3%) 46 (36,8%***) 59 (53,6%***)
Semaine 52 - 25 (43,1%) 23 (56,1%) - - -
Disparition de la
dactylite n (%)†
Semaine 16 10 (37%) 21 (65,6%*) 26 (56,5%) 40 (32,3%) 46 (57,5%***) 54 (65,9%***)
Semaine 52 - 21 (65,6%) 32 (69,6%) NA NA NA
Disparition de
l'enthésite n (%)‡
Semaine 16 17 (26,2%) 32 (50,0%**) 32 (57,1%***) 68 (35,4%) 77 (54,6%***) 78 (55,7%***)
Semaine 52 - 31 (48,4%) 30 (53,6%) NA NA NA
* p < 0,05, ** p <
0,01, *** p <
0,001; par rapport
au placebo Toutes
les valeurs p sont
présentées sans
correction pour les
tests multiples.
Les patients dont
les critères d'évalu
ation binaires
étaient manquants
ont été enregistrés
comme non répondeurs
( "non-responder
imputation" ). NA:
non disponible (Not
Available); ACR:
American College of
Rheumatology; PASI:
Psoriasis Area and
Severity Index;
DAS: score d'activit
é de la maladie
(Disease Activity
Score); BSA: surface
corporelle (Body
Surface Area, BSA)
◊ Critère d'évaluati
on principal 1
Cosentyx 150 mg ou
300 mg SC aux
semaines 0, 1, 2, 3
et 4, puis la même
dose chaque mois. †
Pour les patients
présentant une
dactylite à l'inclus
ion (n = 27, 32 et
46 respectivement
dans PsA2 et n =
124, 80 et 82
respectivement dans
PsA3), la disparitio
n complète de la
dactylite a été
évaluée dans le
sous-groupe de
patients présentant
une dactylite à
l'inclusion et elle
est exprimée en
pourcentage de
patients avec une
valeur de l'indice
de dactylite de
Leeds (LDI) égale à
zéro. ‡ Pour les
patients présentant
une enthésite à
l'inclusion (n =
65, 64 et 56 respect
ivement dans PsA2
et n = 192, 141 et
140 respectivement
dans PsA3), la
disparition complète
de l'enthésite a
été évaluée dans le
sous-groupe de
patients présentant
une enthésite à
l'inclusion et elle
est exprimée en
pourcentage de
patients avec une
valeur de l'indice
d'enthésite de
Leeds (LEI) égale à
zéro.
L'effet de Cosentyx est apparu à la semaine 2. Une différence statistiquement significative pour l'ACR 20 par rapport au placebo a été obtenue à la semaine 3. Dans l'étude PsA2, l'efficacité a été maintenue jusqu'à la semaine 104 (64,4% et 69,4% pour 150 mg et 300 mg, respectivement).
À la semaine 16, les patients traités par Cosentyx ont présenté des améliorations significatives des signes et symptômes, notamment une réponse significativement plus élevée en termes d'ACR 20 (60,0% et 57,0% pour 150 mg et 300 mg, respectivement) par rapport au placebo (18,4%).
Le pourcentage de patients ayant présenté une réponse ACR 20 par visite est présenté à la figure 1
Figure 1: Réponse ACR 20 dans l'étude PsA2 au cours du temps jusqu'à la semaine 24
Pour les critères d’évaluation principaux et les critères d’évaluation secondaires importants, une réponse similaire a été observée chez les patients atteints de RP, qu’ils reçoivent ou non du MTX en même temps.
Les patients non traités antérieurement par un anti-TNF-alpha, mais également les patients anti-TNFalpha-IR qui ont reçu Cosentyx ont présenté une réponse ACR 20 significativement plus élevée aux semaines 16 et 24 par rapport au placebo, la réponse étant numériquement plus élevée dans le groupe non antérieurement traité par anti-TNF-alpha (non traité par anti-TNF-alpha dans PsA2: 64% et 58% pour 150 mg et 300 mg, respectivement, par rapport à 15,9% pour le placebo; anti-TNFalpha-IR: 30% et 46% pour 150 mg et 300 mg, respectivement, par rapport à 14,3% pour le placebo).
Les patients anti-TNF-alpha-IR qui ont été traités par une dose de 300 mg ont présenté un taux de réponse plus élevé selon l'ACR20 (p < 0,05) par rapport aux patients sous placebo et ont montré un bénéfice cliniquement significatif par rapport à la dose de 150 mg pour l'ACR 50, le PASI 75, le PASI 90, l'HAQ-DI, la dactylite et l'enthésite.
Dans PsA2, la proportion de patients ayant obtenu une réponse modifiée selon les critères de réponse au RP (PsARC) était plus élevée à la semaine 24 dans le groupe de patients traités par Cosentyx (59,0% et 61,0% pour 150 mg et 300 mg, respectivement) que dans le groupe de patients traités par placebo (26,5%).
Les résultats des composantes des critères ACR de réponse sont présentés dans le tableau 8.
Tableau 8: Différence des moyennes des composantes de l'ACR par rapport à l'inclusion dans l'étude PsA2 à la semaine 24
Placebo (N = 98) 150 mg (N = 100) 300 mg (N = 100)
Nombre d'articulations gonflées
Inclusion 12,1 11,9 11,2
Différence -5,14 -6,32 -7,28*
Nombre d'articulations sensibles
à la pression
Inclusion 23,4 24,1 20,2
Différence -4,28 -11,42*** -10,84**
Évaluation de la douleur par le
patient
Inclusion 55,4 58,9 57,7
Différence -11,71 -23,39** -22,35**
Évaluation globale par le patient
Inclusion 57,6 62,0 60,7
Différence -10,14 -25,78*** -26,70***
Évaluation globale par le médecin
Inclusion 55,0 56,7 55,0
Différence -25,23 -32,97* -38,52***
Indice de handicap (HAQ)
Inclusion 1,1684 1,2200 1,2828
Différence -0,31 -0,48* -0,56**
CRP (mg/dl)
Inclusion 7,71 14,15 10,69
hsCRP, (rapport post-inclusion/inc 0,75 0,55* 0,55*
lusion)
* p < 0,05, ** p < 0,01, *** p <
0,001 basé sur une valeur p
nominale mais non ajustée
Dans l'étude PsA1, les patients traités par Cosentyx ont présenté des signes et symptômes de RP significativement améliorés à la semaine 24, avec une réponse similaire à celle de l'étude PsA2. L'efficacité a été maintenue jusqu'à la semaine 104.
Réponse radiographique
Dans l'étude PsA3, les atteintes structurales ont été évaluées par radiographie et exprimées sous la forme du score total de Sharp modifié (mTSS) et de ses composantes, le score d'érosion (ES) et le score de pincement articulaire (JSN). Des radiographies des mains, des poignets et des pieds ont été réalisées à l'inclusion, à la semaine 16 et/ou à la semaine 24, et évaluées indépendamment par au moins deux évaluateurs, qui étaient en situation d'aveugle quant au groupe de traitement et au numéro de la visite.
Le traitement par Cosentyx 150 mg ou 300 mg a permis de réduire significativement, par rapport au traitement par placebo, le taux de progression des lésions articulaires périphériques, évalué par la variation du mTSS à la semaine 24 par rapport à l'inclusion (tableau 8).
Le pourcentage de patients sans progression de la maladie (définie par une modification du mTSS ≤0,5) entre la randomisation et la semaine 24 était respectivement de 79,8%, 88,0% et 73,6% pour Cosentyx 150 mg, 300 mg et le placebo. Une inhibition des atteintes structurales a été constatée indépendamment de l'utilisation concomitante éventuelle de MTX ou du statut TNF.
Le traitement par Cosentyx 150 mg a permis de réduire significativement le taux de progression des lésions articulaires périphériques jusqu'à la semaine 24 par rapport au traitement par placebo, d'après la variation du mTSS par rapport à l'inclusion (voir tableau 9). L'inhibition des atteintes structurales s'est maintenue sous traitement par Cosentyx jusqu'à la semaine 52.
Tableau 9: Modification du score total de Sharp modifié dans les études PsA3 et PsA1
PsA3
Placebo n = 296 150 mg1 n = 213 300 mg1 n = 217
Score total
Inclusion 15,0 13,6 12,9
(ET) (38,2) (25,9) (23,7)
Modification moyenne à la semaine 0,5 0,17* 0,08*
24
* p < 0,05, sur la base de la
valeur p nominale non corrigée
pour les tests multiples.
1Cosentyx 150 mg ou 300 mg SC aux
semaines 0, 1, 2, 3 et 4, puis la
même dose chaque mois.
Manifestations axiales dans le RP
Une étude randomisée, en double aveugle et contrôlée par placebo (MAXIMISE) a évalué l'efficacité du sécukinumab chez 485 patients atteints de RP avec des manifestations axiales, qui n'avaient pas été prétraités par des médicaments biologiques et qui ne répondaient pas suffisamment aux anti-inflammatoires non stéroïdiens (AINS). La variable principale, une amélioration d'au moins 20% des critères ASAS 20 (Assessment of SpondyloArthritis International Society, ASAS) à la semaine 12 a été atteinte (voir tableau 10).
Tableau 10: Réponse clinique dans l'étude MAXIMISE à la semaine 12
Placebo (n = 164) 150 mg (n = 157) 300 mg (n = 164)
Réponse ASAS 20, % (IC à 95%) 31,2 (24,6; 38,7) 66,3 (58,4; 73,3)* 62,9 (55,2; 70,0)*
* p < 0,0001; par rapport au
placebo en utilisant l'imputation
multiple. ASAS: critères de
l'Assessment of SpondyloArthritis
International Society.
Une amélioration de la réponse ASAS 20 pour les deux doses de sécukinumab a été observée à la semaine 4 et s'est maintenue jusqu'à 52 semaines.
Fonction physique et qualité de vie liée à la santé
Dans l'étude PsA2 et l'étude PsA3, les patients traités par Cosentyx 150 mg et 300 mg ont présenté une amélioration de la fonction physique à la semaine 24 et à la semaine 16, respectivement, par rapport aux patients traités par placebo, mesurée par le Health Assessment Questionnaire - Disability Index (HAQ-DI). Les améliorations des résultats aux questionnaires HAQ-DI ont été observées indépendamment de toute exposition antérieure aux anti-TNF-alpha. Des résultats comparables ont été observés dans l'étude PsA1.
Les patients traités par Cosentyx ont rapporté des améliorations visibles de la qualité de vie liée à la santé mesurée par le Short Form (36) Health Survey - Physical Component Summary (SF-36 PCS) (p < 0,001). Il y a également eu des améliorations statistiquement significatives du score FACIT-F (Functional Assessment of Chronic Illness Therapy - Fatigue) pour 150 mg et 300 mg par rapport au placebo à la semaine 24 (p < 0,01), et ces améliorations ont été maintenues dans l'étude PsA2 jusqu'à la semaine 104.
Escalade de dose à 300 mg chez les patients dont la réponse à 150 mg n'est pas satisfaisante.
Les études PsA1, PsA2 et PsA4 ont fourni des données évaluables concernant l'escalade de dose de 150 mg à 300 mg chez 159 patients dont la réponse était non satisfaisante après 92 à 156 semaines. Cela correspond à 21,1% des patients restant dans les études. Après l'escalade de la dose de sécukinumab de 150 mg à 300 mg, une amélioration des taux de réponse ACR et PASI et une réduction de la proportion de patients non répondeurs (< ACR 20; < PASI 75) ont été observées.
Spondylarthrite axiale (axSpA)
Spondylarthrite ankylosante (maladie de Bechterew)
La sécurité et l'efficacité de Cosentyx ont été démontrées dans deux études de phase III randomisées, en double aveugle et contrôlées contre placebo, menées chez 590 patients qui présentaient une spondylarthrite ankylosante (SA) active avec une activité de la maladie définie au moyen de l'indice Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 malgré un traitement par anti-inflammatoires non stéroïdiens (AINS), corticostéroïdes ou médicaments antirhumatismaux modificateurs de la maladie (DMARD). Le diagnostic de SA avait été posé en moyenne depuis 2,7 à 5,8 ans chez les patients de ces études. Les patients présentant une ankylose complète étaient exclus.
Pour les deux études, le critère d'évaluation principal était une amélioration d'au moins 20% selon les critères de l'Assessment of SpondyloArthritis International Society (réponse ASAS 20) à la semaine 16.
Dans l'étude 1 portant sur la spondylarthrite ankylosante (étude AS1) ou l'étude 2 portant sur la spondylarthrite ankylosante (étude AS2), respectivement 27,0% et 38,8% des patients avaient été précédemment traités par des médicaments anti-TNF-alpha, ce traitement ayant été interrompu soit en raison d'une absence de réponse, soit en raison d'une intolérance (patients anti-TNF-alpha-IR).
L'étude AS1 (MEASURE 1) a évalué 371 patients; 14,8%, 33,4% et 13,5% d'entre eux ont reçu respectivement du MTX, de la sulfasalazine et des corticostéroïdes de manière concomitante. Les patients randomisés dans le groupe Cosentyx ont reçu 10 mg/kg par voie IV aux semaines 0, 2 et 4, suivis de 75 mg ou 150 mg SC une fois par mois. Les patients randomisés pour recevoir un placebo et qui n'avaient pas répondu au traitement à la semaine 16, ainsi que tous les patients encore sous placebo à la semaine 24, sont passés au traitement par Cosentyx 75 mg ou 150 mg SC une fois par mois.
L'étude AS2 (MEASURE 2) a évalué 219 patients, dont 11,9%, 14,2% et 8,2% étaient respectivement traités de manière concomitante par MTX, sulfasalazine et corticostéroïdes. Les patients randomisés dans le groupe Cosentyx ont reçu 75 mg ou 150 mg SC aux semaines 0, 1, 2, 3 et 4, puis la même dose chaque mois. Les patients randomisés pour recevoir un placebo à l'inclusion ont été re-randomisés à la semaine 16 pour recevoir Cosentyx SC tous les mois (soit 75 mg, soit 150 mg).
Réponse clinique
Signes et symptômes
Dans l'étude AS2, le traitement par Cosentyx 150 mg est celui qui a entraîné la plus nette amélioration des mesures de l'activité de la maladie par rapport au placebo à la semaine 16 (voir tableau 11).
Tableau 11: Réponse clinique dans l'étude AS2 à la semaine 16
Résultat (valeur p par rapport au Placebo (n = 74) 75 mg (n = 73) 150 mg (n = 72)
placebo)
Efficacité à la semaine 16
Réponse ASAS 20, % 28,4 41,1 61,1***
Réponse ASAS 40, % 10,8 26,0 36,1***
hsCRP, (rapport post-inclusion/inc 1,13 0,61 0,55***
lusion)#
ASAS 5/6, % 8,1 34,2 43,1***
BASDAI, variation moyenne des LS –0,85 –1,92 –2,19***
par rapport à la valeur à
l'inclusion†
ASAS, rémission partielle, % 4,1 15,1 13,9
BASDAI 50, % 10,8 24,7* 30,6**
ASDAS-CRP "major improvement" ˆ 4,1 15,1* 25,0***
* p < 0,05; ** p < 0,01; *** p <
0,001 par rapport au placebo
Toutes les valeurs p ont été
ajustées pour la multiplicité des
tests basés sur une hiérarchie
prédéfinie, à l'exception du
BASDAI 50 et de l'ASDAS-CRP. Les
patients qui n'ont pas répondu
ont été comptabilisés pour le
critère d'évaluation binaire
manquant. ASAS: critères de
l'Assessment of SpondyloArthritis
International Society; BASDAI:
indice d'activité de la
spondylarthrite ankylosante de
Bath; hsCRP: protéine C-réactive
hautement sensible; ASDA: score
d'activité de la spondylarthrite
ankylosante; LS: moindres carrés
(least square) # les valeurs
moyennes de la hsCRP à
l'inclusion sont respectivement
8,30, 5,30 et 8,40 † Les valeurs
moyennes du BASDAI à l'inclusion
sont respectivement de 6,78, 6,57
et 6,59 ˆ ASDAS-CRP "major
improvement" défini comme
amélioration ≥2 unités
À la semaine 16, le traitement par Cosentyx 150 mg a entraîné des améliorations cliniquement pertinentes par rapport à l'inclusion pour chaque composante des critères de réponse ASAS 20 (évaluation globale par le patient, douleur rachidienne totale, BASFI et inflammation du rachis).
Dans l'étude AS2, le traitement par Cosentyx 150 mg a entraîné une amélioration significative de l'ASAS 20 dès 1 semaine après le début du traitement par rapport au placebo. Le pourcentage de patients ayant présenté une réponse ASAS 20 par visite est présenté à la figure 2.
Figure 2: Réponse ASAS 20 dans l'étude AS2 jusqu'à la semaine 16
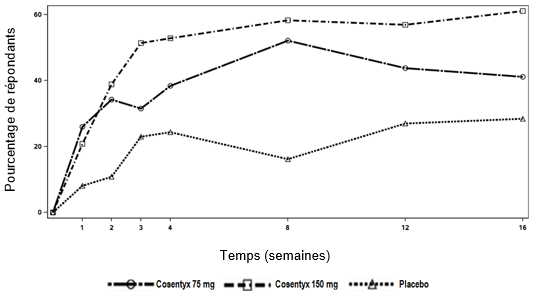
La réponse ASAS 20 était supérieure à celle du placebo à la semaine 16, tant chez les patients n'ayant jamais reçu d'anti-TNF-alpha auparavant (68,2% contre 31,1%; p < 0,05) que chez les patients anti-TNF-alpha-IR (50,0% contre 24,1%; p < 0,05) avec Cosentyx 150 mg. La réponse ASAS 20 en termes de statut HLA B27 a été analysée dans l'étude AS2. 57/72 (79%) patients randomisés pour recevoir 150 mg et 58/74 (78%) patients randomisés pour recevoir un placebo étaient HLA B27 positifs. Pour 3 patients du groupe 150 mg et 5 patients du groupe placebo, le statut HLA B27 n'était pas connu.
Parmi les patients dont les mesures ASAS 20 étaient disponibles, l'ASAS 20 pour la cohorte de 150 mg à la semaine 16 était de 37/54 (68,5%) pour les patients HLA B27 positifs et de 5/9 (55,6%) pour les patients HLA B27 négatifs. Pour le placebo, l'ASAS 20 à la semaine 16 était de 18/51 (35,3%) pour les patients HLA B27 positifs et de 3/8 (37,5%) pour les patients HLA B27 négatifs.
Dans les deux études, la réponse s'est maintenue dans un ordre de grandeur comparable sous Cosentyx/- SensoReady jusqu'à la semaine 52. Sur les 72 patients randomisés pour recevoir Cosentyx 150 mg dans l'étude AS2, 61 (84,7%) étaient toujours sous traitement à la semaine 52. Respectivement 45 et 35 patients présentaient une réponse ASAS 20/40.
Mobilité de la colonne vertébrale:
La mobilité de la colonne vertébrale a été évaluée à l'aide de l'index BASMI (Bath Ankylosing Spondylitis Metrology Index) jusqu'à la semaine 52. Dans l'étude AS2 (150 mg) et l'étude AS1 (75 mg et 150 mg), des améliorations numériques ont été mises en évidence dans chacune des composantes de l'index BASMI pour les patients traités par Cosentyx par rapport aux patients traités par placebo aux semaines 4, 8, 12 et 16 (à l'exception de la flexion lombaire latérale aux semaines 4, 8 et 12 chez les patients traités par une dose de 75 mg après la dose initiale intraveineuse).
Fonction physique et qualité de vie liée à la santé
Dans les études AS1 et AS2, les patients traités par Cosentyx 150 mg ont montré une amélioration de la qualité de vie liée à la santé, mesurée par le questionnaire ASQoL (Ankylosing Spondylitis Quality of Life) (p < 0,001) et le questionnaire SF-36 Physical Component Summary (SF-36 PCS) (p < 0,001). Les patients traités par Cosentyx 150 mg ont également montré des améliorations significatives par rapport au placebo en ce qui concerne les critères d'évaluation exploratoires concernant la fonction physique mesurée par l'indice BASFI (indice fonctionnel de la spondylarthrite ankylosante de Bath [Bath Ankylosing Spondylitis Functional Index]), la douleur mesurée à l'aide de l'échelle de dorsalgie globale et nocturne (Total and Nocturnal Back Pain) et l'épuisement mesuré à l'aide de l'échelle FACIT-F (Functional Assessment of Chronic Illness Therapy - Fatigue). Toutes ces améliorations des fonctions corporelles ont été maintenues jusqu'à la semaine 52.
Spondylarthrite axiale non radiographique (nr-axSpA)
La sécurité et l'efficacité du sécukinumab ont été évaluées dans le cadre d'une étude de phase III randomisée, en double aveugle et contrôlée par placebo, menée chez 555 patients atteints de spondylarthrite axiale non radiographique (nr-axSpA) active qui répondaient aux critères de classification de l'ASAS (Assessment of SpondyloArthritis international Society) pour la spondyloarthrite axiale (axSpA) sans preuve radiographique de modifications des articulations sacro-iliaques, qui auraient répondu aux critères de New York modifiés pour la spondylarthrite ankylosante (AS). Les patients participant à l'étude présentaient une activité de la maladie définie par le Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4, un degré de douleur sur l'échelle visuelle analogique (EVA) pour les dorsalgies totales ≥40 (sur une échelle de 0 à 100 mm), malgré un traitement actuel ou antérieur par des anti-inflammatoires non stéroïdiens (AINS), un taux élevé de protéine C-réactive (CRP) et/ou des signes de sacro-iliite à l'imagerie par résonance magnétique (IRM). Chez les patients de cette étude, le diagnostic d'axSpA avait été posé en moyenne 2,1 à 3,0 ans auparavant et 54% des participants à l'étude étaient des femmes.
Dans l'étude nr-axSpA 1, 9,7% des patients avaient déjà été traités auparavant par un inhibiteur du TNF-α, qui avait été arrêté soit en raison d'un manque d'efficacité, soit en raison d'une intolérance (patients anti-TNFα-IR).
L'étude nr-axSpA 1 (PREVENT) a évalué 555 patients, dont 9,9% et 14,8% recevaient simultanément du MTX ou de la sulfasalazine, respectivement. Au cours de la phase en double aveugle, les patients ont reçu soit un placebo, soit le sécukinumab pendant 52 semaines. Les patients randomisés pour recevoir du sécukinumab ont reçu 150 mg par voie sous-cutanée aux semaines 0, 1, 2, 3 et 4, puis la même dose à intervalles mensuels ou une injection mensuelle de 150 mg de sécukinumab. Le critère d'évaluation principal consistait en une amélioration d'au moins 40% selon les critères de l'Assessment of SpondyloArthritis International Society (réponse ASAS 40) à la semaine 16 chez les patients naïfs de TNFα.
Réponse clinique
Signes et symptômes:
Dans l'étude nr-axSpA 1, le traitement par 150 mg de sécukinumab a entraîné à la semaine 16 une amélioration plus significative des paramètres d'activité de la maladie par rapport au placebo. Ces paramètres comprennent la réponse ASAS 40, ASAS 5/6, BASDAI, BASDAI 50, la CRP haute sensibilité (hsCRP), la réponse ASAS 20 et la rémission partielle ASAS par rapport au placebo à la semaine 16 (tableau 12).
Tableau 12: Réponse clinique dans l'étude nr-axSpA 1 à la semaine 16
Résultat (valeur p par rapport au placebo) Placebo 150 mg1
Nombre de patients naïfs d'anti-TNFα randomisés 171 164
Réponse ASAS 40, % 29,2% 41,5%*
Nombre total de patients randomisés 186 185
Réponse ASAS 40, % 28,0% 40,0%*
ASAS 5/6, % 23,7% 40,0%**
BASDAI, variation moyenne des LS par rapport à -1,46 -2,35**
l'inclusion
BASDAI 50, % 21,0% 37,3%**
hsCRP, (rapport post-inclusion/inclusion) 0,91 0,64**
Réponse ASAS 20, % 45,7% 56,8%*
Rémission partielle ASAS, % 7,0% 21,6%**
* p < 0,05; ** p < 0,001 par rapport au placebo Toutes
les valeurs p ont été soumises à un ajustement pour les
tests multiples sur la base d'une hiérarchie
prédéfinie. En l'absence de données pour un critère
d'évaluation binaire, une imputation en tant que non
répondeur a été effectuée. 1 Sécukinumab 150 mg SC aux
semaines 0, 1, 2, 3 et 4, puis la même dose à
intervalles mensuels ASAS: critères de l'Assessment of
SpondyloArthritis International Society; BASDAI: indice
d'activité de la maladie de la spondylarthrite
ankylosante de Bath; hsCRP: protéine C-réactive
hautement sensible; LS: moindres carrés (least square)
Dans une analyse exploratoire, les patients qui présentaient au début de l'étude à la fois un taux de CRP anormal et des signes d'inflammation à l'IRM sont ceux qui ont le plus bénéficié du sécukinumab.
L'effet du sécukinumab 150 mg s'est manifesté dès la semaine 3 (supérieur au placebo) dans l'étude nr-axSpA 1 chez les patients naïfs d'anti-TNFα en termes de réponse ASAS 40. Le pourcentage de patients qui obtiennent une réponse ASAS 40 parmi les patients naïfs d'anti-TNFα est représenté à la figure 3 au fil des dates de visite. Les patients traités par le sécukinumab ont continué à présenter une réponse jusqu'à la semaine 52 par rapport au placebo.
Figure 3: Réponse ASAS 40 des patients naïfs d'anti-TNFα dans l'étude nr-axSpA 1 au fil du temps jusqu'à la semaine 16
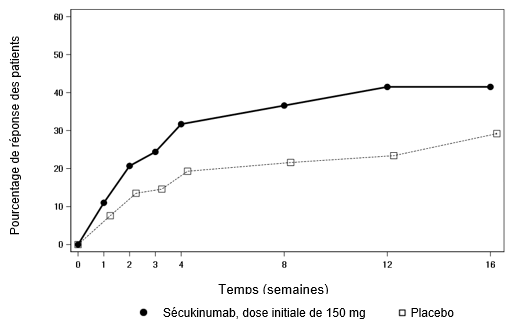
Dans une analyse exploratoire portant sur 54 patients précédemment traités par anti-TNF, une réponse a été observée à la semaine 16 chez 6 patients sur 21 sous Cosentyx et chez 2 patients sur 15 sous placebo.
Fonction physique et qualité de vie liée à la santé:
Les patients traités par le sécukinumab 150 mg ont montré des améliorations statistiquement significatives par rapport aux patients traités par placebo en termes de fonction physique, telle qu'évaluée par BASFI, jusqu'à la semaine 16 (semaine 16: -1,75 contre -1,01, p < 0,01). Par rapport aux patients traités par placebo, les patients traités par le sécukinumab ont rapporté à la semaine 16 des améliorations significatives de la qualité de vie liée à la santé, évaluée à l'aide des questionnaires ASQoL (variation de la moyenne des LS: semaine 16: -3,45 contre -1,84, p < 0,001) et du score cumulé des composantes physiques du SF-36 (SF-36 PCS) (variation de la moyenne des LS: semaine 16: 5,71 contre 2,93, p < 0,001). Ces améliorations ont été maintenues jusqu'à la semaine 52.
Inhibition de l'inflammation à l'imagerie par résonance magnétique (IRM):
Les signes d'inflammation ont été évalués par IRM à l'inclusion et à la semaine 16 et exprimés sous la forme d'une modification du score d'œdème de l'articulation SI de Berlin pour les articulations sacro-iliaques, et du score ASspiMRI-a et du score de la colonne vertébrale de Berlin pour la colonne vertébrale. Une inhibition des signes d'inflammation a été observée chez les patients traités par le sécukinumab, tant au niveau de l'articulation sacro-iliaque que de la colonne vertébrale. La variation moyenne par rapport à l'inclusion du score d'œdème de l'articulation SI de Berlin était de -1,68 chez les patients traités par le sécukinumab 150 mg (n = 180) contre -0,39 chez les patients traités par placebo (n = 174) (p < 0,0001).
Arthrite juvénile idiopathique (AJI)
Arthrite liée à l'enthésite (ERA) et rhumatisme psoriasique juvénile (RPJ)
L'efficacité et la sécurité du sécukinumab ont été examinées dans une étude de phase III randomisée, basée sur les événements, contrôlée contre placebo, en double aveugle et en trois parties, menée chez 86 patients âgés de 2 ans à < 18 ans atteints d'ERA ou de RPJ actifs, qui avaient été diagnostiqués selon les critères modifiés pour la classification des AJI de l'International League of Associations for Rheumatology (ILAR). L'étude se composait d'une partie en ouvert (partie 1), suivie d'un sevrage randomisé (partie 2) et enfin d'un traitement en ouvert (partie 3). Les sous-groupes de patients atteints d'AJI au début de l'étude étaient les suivants: 60,5% ERA et 39,5% RPJ.
À l'inclusion, 65,1% des patients avaient reçu du MTX (63,5% (33 sur 52) des patients atteints d'ERA et 67,6% (23 sur 34) des patients atteints de RPJ). Les patients ont reçu une dose de 75 mg lorsqu'ils pesaient < 50 kg, ou de 150 mg lorsqu'ils pesaient ≥50 kg. Le critère d'évaluation principal était le délai jusqu'à une poussée de la maladie dans la partie 2. Une poussée de la maladie était définie comme une dégradation de ≥30% dans au moins trois des six critères de réponse ACR pour l'AJI et une amélioration de ≥30% dans pas plus d'un des six critères de réponse ACR pour l'AJI.
Dans la partie 1 en ouvert de l'étude, tous les patients ont reçu du sécukinumab jusqu'à la semaine 12. Les patients qui avaient été classés comme répondeurs en semaine 12 ont intégré la phase en double aveugle de la partie 2 et ont été randomisés selon un rapport de 1:1 soit pour poursuivre le traitement par sécukinumab, soit pour commencer un traitement par placebo. À la fin de la partie 1, 75 des 86 patients (90,4%) présentaient une réponse ACR 30 pour l'AJI et ont pris part à la partie 2.
L'étude a atteint son critère d'évaluation principal en démontrant un allongement statistiquement significatif du délai jusqu'à une poussée de la maladie chez les patients traités par sécukinumab par rapport au placebo. Le risque d'une poussée de la maladie a été réduit de 72% chez les patients ayant reçu du sécukinumab par rapport aux patients ayant reçu le placebo (hazard ratio des poussées de la maladie = 0,28, IC à 95%: 0,13 à 0,63, p < 0,001) (figure 4). Dans la deuxième partie de l'étude, des poussées ont été observées chez 21 patients au total dans le groupe placebo (11 RPJ et 10 ERA) contre 10 patients dans le groupe sous sécukinumab (4 RPJ et 6 ERA). L'analyse de survie du délai jusqu'à la survenue d'une poussée de la maladie a montré que le traitement par sécukinumab a conduit, par rapport au groupe placebo de la partie 2, à un allongement du délai jusqu'à la survenue d'une poussée de la maladie, indépendamment de la prise de MTX à l'inclusion. Le risque d'une poussée de la maladie était réduit de 35% chez les sujets sous sécukinumab avec traitement simultané par MTX (hazard ratio = 0,65, IC à 95%: 0,23, 1,83) et de 92% chez les sujets sous sécukinumab sans traitement simultané par MTX (hazard ratio = 0,08, IC à 95%: 0,02, 0,32), par rapport aux sujets qui avaient été randomisés dans le groupe placebo dans la partie 2.
Figure 4: Estimations de Kaplan-Meier du délai jusqu'à la poussée de la maladie dans la partie 2
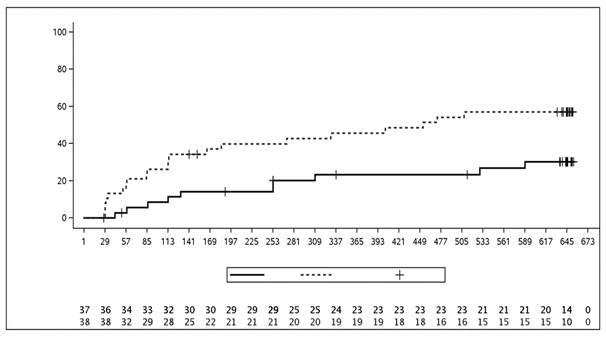
Hidrosadénite suppurée
La sécurité et l'efficacité du sécukinumab ont été évaluées chez 1084 patients adultes atteints d'hidrosadénite suppurée (HS) modérée à sévère et éligibles à un traitement systémique par des médicaments biologiques, dans deux études de phase III randomisées, en double aveugle et contrôlées contre placebo. Les patients inclus dans l'étude HS 1 (SUNSHINE) ou dans l'étude HS 2 (SUNRISE) présentaient un stade I de Hurley (4,6% et 2,8% resp.), II (61,4% et 56,7% resp.) ou III (34,0% et 40,5% resp.) à l'inclusion et présentaient au moins cinq lésions inflammatoires affectant deux régions anatomiques. La proportion de patients ayant un poids corporel ≥90 kg était de 54,7% dans l'étude HS 1 et de 50,8% dans l'étude HS 2. Les patients inclus dans ces études présentaient un diagnostic d'HS modérée à sévère depuis 7,3 ans en moyenne, et 56,3% des participants à l'étude étaient des femmes. Dans l'étude HS 1 et l'étude HS 2, 82,3% et 83,6% des patients avaient été précédemment traités par des antibiotiques systémiques et 23,8% et 23,2% des patients avaient été précédemment traités par un médicament biologique qu'ils avaient arrêté, soit en raison d'une efficacité insuffisante, soit en raison d'une intolérance (patients exposés à un médicament biologique).
L'étude HS 1 a porté sur 541 patients et l'étude HS 2 sur 543 patients, dont respectivement 12,8% et 10,7% ont reçu simultanément une dose constante d'antibiotiques. Dans les deux études, les patients randomisés pour recevoir le sécukinumab ont reçu 300 mg de sécukinumab par voie sous-cutanée aux semaines 0, 1, 2, 3 et 4, puis 300 mg toutes les 2 semaines (Q2W) ou toutes les 4 semaines (Q4W). À la semaine 16, les patients randomisés pour recevoir le placebo ont été réaffectés et ont reçu 300 mg de sécukinumab à chacune des semaines 16, 17, 18, 19 et 20, puis soit 300 mg de sécukinumab Q2W, soit 300 mg de sécukinumab Q4W.
Le critère d'évaluation principal dans les deux études (étude HS 1 et étude HS 2) était la proportion de patients ayant obtenu une réponse clinique de l'hidrosadénite suppurée à la semaine 16. Celle-ci était définie par une diminution d'au moins 50% du nombre total d'abcès et de nodules inflammatoires et par l'absence d'augmentation du nombre d'abcès et/ou du nombre de fistules drainantes par rapport à l'inclusion (HiSCR50). La diminution des douleurs cutanées liées à l'HS a été évaluée en tant que critère d'évaluation secondaire à partir des données regroupées des études HS 1 et HS 2 à l'aide d'une échelle d'évaluation numérique (NRS [Numeric Rating Scale]) chez les patients présentant un score initial égal ou supérieur à 3 lors de l'inclusion.
Dans l'étude HS 1 et l'étude HS 2, une proportion significativement plus élevée de patients traités par 300 mg de sécukinumab Q2W a obtenu une réponse HiSCR50 à la semaine 16, avec une diminution significative du nombre d'abcès et de nodules inflammatoires (AN) par rapport au placebo, ce qui n'a cependant pas été le cas des patients traités par 300 mg de sécukinumab Q4W par rapport au placebo dans l'étude HS 1. Dans l'étude HS 2, une différence statistiquement significative a également été observée dans la réponse HiSCR50 et le nombre d'AN avec le traitement sécukinumab 300 mg Q4W par rapport au placebo. Dans le groupe sécukinumab 300 mg Q4W de l'étude HS 2 ainsi que dans le groupe sécukinumab 300 mg Q2W de l'étude HS 1, les poussées de la maladie ont été moins nombreuses jusqu'à la semaine 16 par rapport au placebo. Une proportion significativement plus élevée de patients traités par 300 mg de sécukinumab Q2W (données regroupées) a enregistré une diminution cliniquement significative des douleurs cutanées liées à l'HS à la semaine 16 par rapport au placebo.
Tableau 13: Réponse clinique dans l'étude HS 1 et l'étude HS 2 à la semaine 161
Étude HS 1 Étude HS 2
Placebo 300 mg Q4W 300 mg Q2W Placebo 300 mg Q4W 300 mg Q2W
Nombre des patients 180 180 181 183 180 180
randomisés
Réponse HiSCR50, % 33,7 41,8 45,0* 31,2 46,1* 42,3*
Nombre d'AN, % -24,3 -42,4 -46,8* -22,4 -45,5* -39,3*
Moyenne des LS pour
la variation par
rapport à l'inclusio
n
Poussées, % 29,0 23,2 15,4* 27,0 15,6* 20,1
Données regroupées
(étude HS 1 et
étude HS 2)
Placebo 300 mg Q4W 300 mg Q2W
Nombre de patients 251 252 266
présentant une NRS
≥3 à l'inclusion
Réponse NRS 30, % 23,0 33,5 36,6*
1 Une imputation
multiple a été
effectuée en cas de
données manquantes
* Statistiquement
significatif par
rapport au placebo
sur la base de la
hiérarchie prédéfini
e avec un alpha
global = 0,05 AN:
abcès et nodules
inflammatoires;
HiSCR: réponse
clinique à l'hidrosa
dénite; NRS: échelle
d'évaluation
numérique
Dans les deux études, le sécukinumab a commencé à agir dès la semaine 2, son efficacité a augmenté jusqu'à la semaine 16 et s'est maintenue jusqu'à la semaine 52.
Des améliorations ont été observées pour les critères d'évaluation principaux et les critères secondaires importants chez les patients atteints d'HS, indépendamment de tout traitement antibiotique antérieur ou concomitant. La réponse HiSCR50 s'est améliorée à la semaine 16, tant chez les patients naïfs de traitement biologique que chez les patients exposés aux traitements biologiques. Des améliorations plus importantes à la semaine 16 par rapport à la valeur initiale en comparaison avec le placebo ont été mises en évidence en ce qui concerne la qualité de vie liée à la santé, mesurée avec le Dermatology Life Quality Index.
PharmacocinétiqueAbsorption
Les données de cette section sont basées sur une analyse pharmacocinétique de population.
Après l'administration d'une dose unique sous-cutanée de 150 mg ou de 300 mg administrée en 2 injections de 150 mg à des patients atteints de psoriasis en plaques, le sécukinumab a atteint des pics de concentration sérique de 13,7 ± 4,8 µg/ml et 27,3 ± 9,5 µg/ml, respectivement, entre 5 et 6 jours après l'administration de la dose.
Après l'introduction de la dose hebdomadaire pendant le premier mois, le temps nécessaire pour atteindre la concentration maximale était de 31 à 34 jours.
Les pics de concentration à l'état d'équilibre (Cmax,ss) après l'administration sous-cutanée de 150 mg ou de 300 mg étaient respectivement de 27,6 µg/ml et 55,2 µg/ml. Avec les schémas d'administration mensuels, l'état d'équilibre est atteint après 20 semaines.
Après des doses d'entretien mensuelles répétées, les patients ont présenté un doublement de la concentration sérique maximale comparativement à l'exposition après une dose unique.
Le sécukinumab est absorbé avec une biodisponibilité absolue moyenne de 73% après injection sous-cutanée dans l'abdomen. L'absorption du sécukinumab après injection sous-cutanée dans le bras ou la cuisse n'a pas été étudiée.
Chez les patients atteints de psoriasis en plaques, l'exposition systémique au sécukinumab après une injection sous-cutanée unique de 300 mg de solution injectable en seringue préremplie était similaire à l'exposition précédemment observée après deux injections de 150 mg.
Dans l'étude HS 1 et l'étude HS 2, après administration sous-cutanée de 300 mg aux semaines 0, 1, 2, 3 et 4, la concentration résiduelle moyenne à l'état d'équilibre (± ET) du sécukinumab à la semaine 16 était respectivement d'environ 55,1 ± 26,7 µg/ml et 58,1 ± 30,1 µg/ml pour le traitement par 300 mg de sécukinumab toutes les 2 semaines et de respectivement 28,8 ± 18,3 µg/ml et 30,2 ± 18,0 µg/ml pour le traitement par 300 mg de sécukinumab toutes les 4 semaines.
Distribution
Le volume de distribution moyen pendant la phase terminale (Vz) après l'administration d'une dose unique par voie intraveineuse était compris entre 7,10 et 8,60 l chez les patients atteints de psoriasis en plaques. Cela indique que le sécukinumab est soumis à une distribution limitée dans les compartiments périphériques.
Une et deux semaines après l'administration d'une dose unique de 300 mg de sécukinumab par voie sous-cutanée (administrée en deux injections de 150 mg), les concentrations de sécukinumab dans le fluide interstitiel cutané des patients atteints de psoriasis en plaques correspondaient à 28% à 39% des concentrations sériques.
Métabolisme
Pas de données.
Élimination
La clairance systémique moyenne était de 0,19 l/j chez les patients atteints de psoriasis en plaques. Comme on pouvait s'y attendre avec un anticorps monoclonal thérapeutique IgG1 qui interagit avec une cytokine cible soluble telle que l'IL-17A, la clairance était indépendante de la dose et du temps.
La demi-vie d'élimination moyenne estimée chez les patients atteints de psoriasis en plaques est de 27 jours. Chez les patients individuels atteints de psoriasis en plaques, la demi-vie estimée se situait entre 17 et 41 jours. Dans une analyse pharmacocinétique de population, la clairance systémique moyenne après administration sous-cutanée de 300 mg aux semaines 0, 1, 2, 3 et 4, puis de 300 mg toutes les 2 semaines ou toutes les 4 semaines, était de 0,26 l/jour chez les patients atteints d'hidrosadénite suppurée.
La demi-vie d'élimination moyenne, estimée à partir d'une analyse pharmacocinétique de population, était de 23 jours chez les patients atteints d'hidrosadénite suppurée.
Linéarité/non-linéarité
La pharmacocinétique (PK) du sécukinumab après l'administration d'une dose unique ainsi qu'après des doses multiples chez des patients atteints de psoriasis en plaques a été déterminée dans plusieurs études avec des doses intraveineuses allant de 1 × 0,3 mg/kg à 3 × 10 mg/kg et des doses sous-cutanées allant de 1 × 25 mg à des doses répétées de 300 mg. L'exposition a été proportionnelle à la dose dans tous les schémas d'administration.
Les propriétés PK du sécukinumab relevées chez des patients atteints de rhumatisme psoriasique et de spondylarthrite axiale (spondylarthrite ankylosante et spondylarthrite axiale non radiographique) étaient similaires à celles observées chez les patients présentant un psoriasis en plaques.
Troubles de la fonction hépatique / troubles de la fonction rénale
Aucune donnée pharmacocinétique n'est disponible chez les patients souffrant d'insuffisance hépatique ou rénale.
Patients âgés
Parmi les 3430 patients atteints de psoriasis en plaques ayant été exposés au sécukinumab dans les études cliniques, 230 patients au total étaient âgés de 65 ans ou plus et 32 patients de 75 ans ou plus. Parmi les 721 patients atteints d'hidrosadénite suppurée ayant été traités par Cosentyx dans les études cliniques, 11 patients au total étaient âgés de 65 ans ou plus et 0 patient était âgé de 75 ans ou plus.
Parmi les 2536 patients atteints de rhumatisme psoriasique ayant été exposés au Cosentyx dans les études cliniques, 236 patients au total étaient âgés de 65 ans ou plus et 25 patients de 75 ans ou plus.
Parmi les 571 patients atteints de spondylarthrite ankylosante ayant été exposés au Cosentyx dans les études cliniques, 24 patients au total étaient âgés de 65 ans ou plus et 3 patients de 75 ans ou plus.
Parmi les 524 patients atteints de spondylarthrite axiale non radiographique qui ont reçu Cosentyx dans les études cliniques, 9 patients étaient âgés de 65 ans ou plus et 2 patients étaient âgés de 75 ans ou plus.
Sur la base d'une analyse pharmacocinétique de population, la clairance chez les patients âgés était comparable à la clairance chez les patients de moins de 65 ans.
Patients pédiatriques
Psoriasis en plaques
Dans un pool des deux études pédiatriques, le sécukinumab a été administré aux patients atteints de psoriasis en plaques modéré à sévère (âgés de 6 à < 18 ans) selon le schéma posologique pédiatrique recommandé. À la semaine 24, les patients dont le poids corporel était ≥25 et < 50 kg présentaient une concentration résiduelle moyenne ± ET à l'état d'équilibre de 19,8 ± 6,96 μg/ml (n = 24) après avoir reçu 75 mg de sécukinumab. Les patients dont le poids corporel était ≥50 kg présentaient une concentration résiduelle moyenne à l'état d'équilibre de 27,3 ± 10,1 μg/ml (n = 36) après avoir reçu 150 mg de sécukinumab. La concentration résiduelle moyenne ± ET à l'état d'équilibre chez les patients dont le poids corporel était < 25 kg (n = 8) était de 32,6 ± 10,8 μg/ml à la semaine 24 après administration de la dose de 75 mg. La biodisponibilité du sécukinumab est environ 26% plus élevée chez les patients pédiatriques que chez les adultes.
Arthrite juvénile idiopathique (AJI): arthrite liée à l'enthésite (ERA) et rhumatisme psoriasique juvénile (RPJ)
Dans une étude pédiatrique, les patients atteints d'ERA et de RPJ (âgés de 2 ans à moins de 18 ans) ont reçu du sécukinumab à la posologie pédiatrique recommandée. À la semaine 24, les patients dont le poids corporel était < 50 kg et les patients dont le poids corporel était ≥50 kg présentaient une concentration résiduelle moyenne ± ET à l'état d'équilibre de respectivement 25,2 ± 5,45 microgrammes/ml (n = 10) et 27,9 ± 9,57 microgrammes/ml (n = 19).
Données précliniquesLes données précliniques basées sur des tests de réactivité tissulaire croisée, des études de pharmacologie de sécurité, des études de toxicité à doses répétées et des études sur la toxicité pour la reproduction réalisées avec le sécukinumab ou un anticorps murin anti-IL-17A de souris n'indiquent aucun risque particulier pour l'homme.
Comme le sécukinumab se lie à l'IL-17A des singes cynomolgus et des humains, sa sécurité a été étudiée chez les singes cynomolgus.
Toxicité à long terme (ou toxicité en cas d'administration répétée)
Aucun effet indésirable n'a été observé après l'administration sous-cutanée de sécukinumab à des singes cynomolgus pendant une durée allant jusqu'à 13 semaines et après l'administration intraveineuse pendant une durée allant jusqu'à 26 semaines [y compris les évaluations de pharmacocinétique, de pharmacodynamique, d'immunogénicité et d'immunotoxicité (par exemple, réponse des anticorps dépendant des cellules T et activité des cellules NK)]. Les concentrations sériques moyennes observées chez les singes après 13 doses sous-cutanées hebdomadaires de 150 mg/kg sont 48 fois plus élevées que les concentrations sériques moyennes prédites qui sont attendues chez les patients atteints de psoriasis traités à la dose clinique la plus élevée. Le facteur d'augmentation de l'exposition est encore plus important si l'on tient compte de la concentration sérique moyenne de l'étude toxicologique intraveineuse de 26 semaines chez les singes cynomolgus. Les anticorps contre le sécukinumab n'ont été détectés que chez un animal sur 101. L'application du sécukinumab sur des tissus humains normaux n'a pas montré de réactivité croisée tissulaire non spécifique.
Mutagénicité et carcinogénicité
Aucune étude préclinique n'a été réalisée pour évaluer le potentiel mutagène ou carcinogène du sécukinumab.
Toxicité pour la reproduction
Administré pendant l'organogenèse et la fin de la gestation, le sécukinumab n'a pas montré de toxicité maternelle, d'embryotoxicité ni de tératogénicité dans une étude sur le développement embryofœtal chez des singes cynomolgus.
Aucun effet indésirable d'un anticorps murin anti-IL-17A de souris n'a été observé dans les études de fertilité et de développement embryonnaire précoce, prénatal et postnatal chez la souris. La dose élevée utilisée dans ces études était supérieure à la dose maximale efficace en termes d'inhibition et d'activité de l'IL-17A.
Remarques particulièresIncompatibilités
Poudre pour solution injectable:
Cosentyx ne doit pas être mélangé avec des médicaments ou des solvants autres que l'eau stérile pour préparations injectables.
Solution injectable (seringue préremplie et stylo prérempli):
Ces médicaments ne doivent pas être mélangés à d'autres médicaments.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention "EXP" sur l'emballage.
Remarques particulières concernant le stockage
Les médicaments doivent être tenus hors de portée des enfants.
Conserver au réfrigérateur (2–8 °C).
Conserver dans l'emballage d'origine pour protéger le contenu de la lumière.
Uniquement pour la seringue préremplie et le stylo prérempli:
Ne pas congeler.
Si nécessaire, Cosentyx/- SensoReady/- UnoReady peut être conservé non réfrigéré à température ambiante (pas au-dessus de 30 °C) pendant une période unique de 4 jours maximum. Après 4 jours de conservation non réfrigérée, la seringue préremplie et le stylo prérempli doivent être jetés.
Poudre pour solution injectable:
La préparation ne contient pas de conservateur. Sa stabilité chimique et physique "in use" a été démontrée pendant 24 heures à 2 à 8 °C. Pour des raisons microbiologiques, la préparation prête à l'emploi doit être utilisée immédiatement après ouverture.
Remarques concernant la manipulation
La poudre se présente sous la forme d'un lyophilisat solide blanc. La solution dans la seringue préremplie ou le stylo prérempli est incolore à légèrement jaunâtre.
Après l'injection, la solution restant dans le flacon, la seringue préremplie ou le stylo prérempli ne doit pas être utilisé(e) et doit être éliminé(e) conformément aux exigences locales. Le flacon, la seringue préremplie ou le stylo prérempli sont à usage unique.
Vous trouverez ci-dessous des instructions d'utilisation détaillées pour la poudre pour solution injectable.
Numéro d’autorisation63295, 65225, 65226, 68324 (Swissmedic)
PrésentationCosentyx poudre pour solution injectable en flacon de 150 mg: 1 [B].
Cosentyx solution injectable en seringue préremplie de 75 mg: 1 [B].
Cosentyx solution injectable en seringue préremplie de 150 mg: 1 et 2 [B].
Cosentyx solution injectable en seringue préremplie de 300 mg: 1 [B].
Cosentyx SensoReady solution injectable en stylo prérempli de 150 mg: 1 et 2 [B].
Cosentyx UnoReady solution injectable en stylo prérempli de 300 mg: 1 [B].
Titulaire de l’autorisationNovartis Pharma Schweiz AG, Risch; domicile: 6343 Rotkreuz
Mise à jour de l’informationMai 2025
Mode d'emploi de Cosentyx, poudre pour solution injectable
Les informations suivantes sont destinées uniquement aux médecins ou aux professionnels de la santé.
Le flacon à usage unique contient 150 mg de Cosentyx à reconstituer avec 1 ml d'eau stérile pour préparations injectables (SWFI). La préparation de la solution pour injection sous-cutanée doit être effectuée sans interruption et dans le respect des conditions d'asepsie. Le temps de préparation entre le moment où le bouchon est percé et la fin de la reconstitution est en moyenne de 20 minutes et ne doit pas dépasser 90 minutes.
Pour préparer Cosentyx, poudre pour solution injectable, veuillez suivre les instructions suivantes:
Instructions pour la reconstitution de Cosentyx, poudre pour solution injectable:
1.Amenez le flacon de Cosentyx, poudre pour solution injectable à température ambiante et assurez-vous que l'eau stérile pour préparations injectables (SWFI) est également à température ambiante.
2.Aspirez un peu plus de 1,0 ml d'eau stérile pour préparations injectables (SWFI) dans une seringue graduée à usage unique de 1 ml et ajustez le volume à 1,0 ml.
3.Retirez le capuchon en plastique du flacon.
4.Insérez l'aiguille de la seringue au centre du bouchon en caoutchouc du flacon contenant le lyophilisat de Cosentyx et reconstituez le lyophilisat en injectant lentement 1,0 ml de SWFI dans le flacon. Le jet de SWFI doit être dirigé directement sur le lyophilisat.
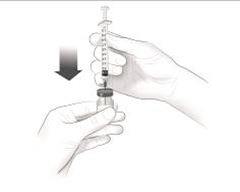
5.Inclinez le flacon à un angle d'environ 45° et tournez-le doucement entre le bout des doigts pendant environ 1 minute. Ne secouez pas et ne retournez pas le flacon.
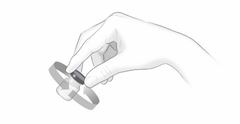
6.Laissez le flacon reposer à température ambiante pendant au moins 10 minutes pour permettre la dissolution du lyophilisat. Il peut y avoir une formation de mousse dans la solution.
7.Inclinez le flacon à un angle d'environ 45° et tournez-le doucement entre le bout des doigts pendant environ 1 minute. Ne secouez pas et ne retournez pas le flacon.
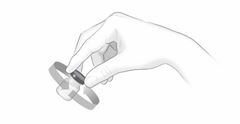
8.Laissez le flacon reposer à température ambiante pendant environ 5 minutes. La solution obtenue doit être transparente. La solution peut être incolore à légèrement jaunâtre. Si la poudre lyophilisée n'est pas complètement dissoute ou si le liquide contient des particules bien visibles, est trouble ou nettement brun, la solution ne doit pas être utilisée.
9.Préparez le nombre de flacons nécessaires (1 flacon de 150 mg pour la dose de 75 mg, 1 flacon de 150 mg pour la dose de 150 mg, 2 flacons de 150 mg pour la dose de 300 mg).
Une fois préparée, la solution pour injection sous-cutanée doit être utilisée immédiatement, car elle ne contient pas de conservateur. Ne pas congeler.
Instructions pour l'administration de la solution Cosentyx
1.Pour aspirer la solution dans la seringue, inclinez le flacon à un angle d'environ 45° et positionnez la pointe de l'aiguille au point le plus bas de la solution dans le flacon. Ne retournez PAS le flacon.
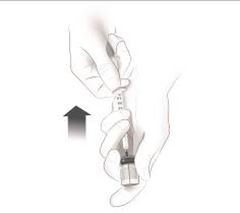
2.Pour les doses de 150 mg et de 300 mg, prélevez avec précaution un peu plus de 1,0 ml de solution pour injection sous-cutanée du flacon dans une seringue graduée à usage unique de 1 ml à l'aide d'une aiguille appropriée (p.ex. 21G × 2"). Cette aiguille est utilisée uniquement pour aspirer Cosentyx dans la seringue à usage unique. Préparez le nombre de seringues nécessaires (1 seringue pour la dose de 150 mg, 2 seringues pour la dose de 300 mg). Pour un enfant recevant la dose de 75 mg, aspirez avec précaution un peu plus de 0,5 ml de la solution pour injection sous-cutanée et jetez immédiatement le reste.
3.Avec l'aiguille dirigée vers le haut, tapotez légèrement sur la seringue pour faire remonter les éventuelles bulles d'air.
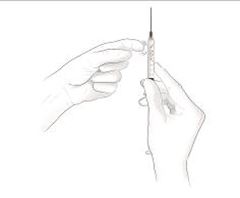
4.Remplacez l'aiguille montée par une aiguille 27G × ½".

5.Expulsez les bulles d'air hors de la seringue. Pour la dose de 150 mg, avancez le piston jusqu'au repère de 1,0 ml. Pour la dose de 75 mg, avancez le piston jusqu'au repère de 0,5 ml.
6.Nettoyez le site d'injection avec un tampon imbibé d'alcool.
7.Le site d'injection recommandé pour l'injection sous-cutanée de la solution Cosentyx est la partie inférieure de l'abdomen (ne pas injecter à moins de 5 cm du nombril).
En variante, on peut également utiliser la face avant de la cuisse ou la face extérieure du bras, si le site d'injection recommandé ne convient pas (voir rubrique Pharmacocinétique).
Choisissez un nouvel endroit pour chaque injection. N'injectez pas la solution à un endroit où la peau est sensible, endommagée ou rouge, ou si elle pèle ou est durcie. Les zones présentant des cicatrices ou des vergetures sont à éviter.
8.La solution restant dans le flacon ne doit pas être utilisée et doit être éliminée conformément aux exigences locales. Les flacons sont à usage unique. Jetez la seringue usagée dans un récipient pour seringues. Les aiguilles et les seringues usagées ne doivent pas être réutilisées.
|