CompositionPrincipes actifs
Empagliflozine, chlorhydrate de metformine.
Excipients
Jardiance Met 5 mg/500mg, Jardiance Met 5 mg/850mg, Jardiance Met 5 mg/1000mg:
-Noyau du comprimé: amidon de maïs, copovidone, silice colloïdale anhydre, stéarate de magnésium
-Pelliculage: hypromellose 2910, macrogol 400, dioxyde de titane (E171), talc, oxyde de fer jaune (E172)
Jardiance Met 12,5 mg/500mg, Jardiance Met 12,5 mg/850mg, Jardiance Met 12,5 mg/1000mg:
-Noyau du comprimé: amidon de maïs, copovidone, silice colloïdale anhydre, stéarate de magnésium
-Pelliculage: hypromellose 2910, macrogol 400, dioxyde de titane (E171), talc, oxyde de fer noir (E172), oxyde de fer rouge (E172)
Forme pharmaceutique et quantité de principe actif par unitéComprimés pelliculés à
5 mg d'empagliflozine et 500 mg de chlorhydrate de metformine ou
5 mg d'empagliflozine et 850 mg de chlorhydrate de metformine ou
5 mg d'empagliflozine et 1000 mg de chlorhydrate de metformine.
12,5 mg d'empagliflozine et 500 mg de chlorhydrate de metformine ou
12,5 mg d'empagliflozine et 850 mg de chlorhydrate de metformine ou
12,5 mg d'empagliflozine et 1000 mg de chlorhydrate de metformine.
Indications/Possibilités d’emploiJardiance Met est indiqué chez les adultes ainsi que chez les enfants et les adolescents à partir de 10 ans pour le traitement du diabète sucré de type 2, en complément d'un régime alimentaire et d'une activité physique, dans les cas suivants:
un contrôle adéquat de la glycémie ne peut être obtenu à la dose maximale tolérée ou recommandée de metformine, en monothérapie ou en association avec d'autres hypoglycémiants (voir rubrique "Efficacité clinique" );
un traitement par l'empagliflozine et la metformine administrées sous forme de comprimés distincts est déjà en cours.
Prévention d'événements cardiovasculaires chez des patients adultes atteints de diabète de type 2 et d'une maladie cardiovasculaire déjà manifeste (voir rubrique "Efficacité clinique" ).
Posologie/Mode d’emploiAdultes:
La dose recommandée doit être déterminée de manière individuelle en se basant sur le traitement actuel, sur l'efficacité et la tolérance chez le patient respectif. Les doses journalières maximales recommandées de 25 mg d'empagliflozine et de 2000 mg de metformine ne doivent pas être dépassées.
-Le traitement par empagliflozine en plus du traitement antérieur doit débuter par Jardiance Met 5 mg d'empagliflozine deux fois par jour (dose journalière 10 mg). La dose de metformine doit correspondre environ à la dose prise dans le cadre du traitement antérieur. Si aucun contrôle satisfaisant de la glycémie n'est obtenu avec la dose de Jardiance Met 5 mg d'empagliflozine deux fois par jour, la dose peut être augmentée à Jardiance Met 12.5 mg d'empagliflozine deux fois par jour (dose journalière 25 mg) chez les patients qui supportent bien une dose plus élevée d'empagliflozine.
-Chez les patients qui sont passés de l'administration de comprimés distincts d'empagliflozine et de metformine à Jardiance Met, la dose journalière d'empagliflozine et de metformine doit correspondre à la dose administrée jusqu'ici ou à la dose thérapeutique adéquate de metformine qui s'en rapproche le plus.
Pour les différentes doses de metformine, Jardiance Met existe aux dosages de 5 mg d'empagliflozine plus 500 mg, 850 mg ou 1000 mg de chlorhydrate de metformine ou 12,5 mg d'empagliflozine plus 500 mg, 850 mg ou 1000 mg de chlorhydrate de metformine.
Jardiance Met doit être pris pendant les repas afin de réduire les effets indésirables gastro-intestinaux dus à la metformine.
Enfants et adolescents:
La dose initiale recommandée d'empagliflozine est de 5 mg deux fois par jour (dose journalière de 10 mg). En cas de bonne tolérance, celle-ci peut être portée à 12,5 mg deux fois par jour (dose journalière de 25 mg) lorsqu'un meilleur contrôle de la glycémie est nécessaire (voir également les informations générales sous "Patients présentant des troubles de la fonction rénale" ). Dans l'étude DINAMO (voir rubrique "Efficacité clinique" ), des doses journalières de 10 mg et 25 mg d'empagliflozine ont été administrées.
La dose journalière maximale recommandée de Jardiance Met est de 25 mg d'empagliflozine et de 2000 mg de metformine.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
L'expérience concernant l'administration d'empagliflozine chez les patients présentant une insuffisance hépatique sévère ou une nette augmentation des transaminases (plus de trois fois supérieure) est fortement limitée. L'utilisation d'empagliflozine n'est pas recommandée chez ces patients.
Patients présentant des troubles de la fonction rénale
Le DFGe doit être évalué avant toute initiation de traitement et au moins une fois par an par la suite. Chez les patients dont l'insuffisance rénale risque de progresser et chez les patients âgés, la fonction rénale doit être évaluée plus fréquemment, par exemple tous les 3 à 6 mois.
Avant toute initiation de traitement par la metformine chez des patients avec un DFGe <60 ml/min/1,73 m2, les facteurs susceptibles d'augmenter le risque d'une acidose lactique (voir "Mises en garde et précautions" ) doivent être évalués.
Tableau 1: Posologie pour les patients insuffisants rénaux
DFGe ml/min/1,73m2 Metformine Empagliflozine
60-89 Une diminution de la dose peut être Pas d'adaptation de la
envisagée selon la détérioration de la posologie. La dose journalière
fonction rénale. La dose journalière maximale chez l'adulte ainsi que
maximale chez l'adulte est de 3000 mg.* La chez l'enfant et l'adolescent
dose journalière maximale chez l'enfant et est de 25 mg.
l'adolescent est de 2000 mg.**
45-59** La dose d'initiation est de 500 mg ou 850 Pas d'adaptation de la posologie.
mg par jour. La dose journalière maximale
est de 1000 mg, répartis en 2 prises
individuelles
30- 44** La dose d'initiation est de 500 mg ou 850 L'empagliflozine n'est pas
mg par jour. La dose journalière maximale recommandée.
est de 1000 mg, répartis en 2 prises
individuelles
< 30** La metformine est contre-indiquée L'empagliflozine n'est pas
recommandée.
* Si aucun dosage adéquat de Jardiance Met n'est disponible, les composants individuels doivent être utilisés à la place de l'association fixe.
** On ne dispose pas de données pour les enfants avec un DFGe <60 ml/min/1,73 m2 ni pour les enfants de moins de 10 ans.
Patients âgés
Étant donné que la fonction rénale peut être limitée chez les patients âgés, la dose de metformine doit être ajustée avec prudence en tenant compte des paramètres de la fonction rénale. Un contrôle régulier de la fonction rénale est nécessaire (voir "Mises en garde et précautions" ).
Enfants et adolescents
Des données sur l'efficacité et la sécurité de l'empagliflozine en association avec la metformine ne sont disponibles que pour le groupe d'âge de 10 à 17 ans (à l'exception des patients avec un DFGe <60 ml/min/1,73 m2) et exclusivement pour l'indication "Traitement du diabète sucré de type 2" . Jardiance Met n'est pas autorisé pour le traitement des enfants de moins de 10 ans, pour lesquels on ne dispose pas de données. L'utilisation de Jardiance Met chez les enfants et les adolescents avec un DFGe <60 ml/min/1,73 m2 n'est pas recommandée, car l'efficacité et la sécurité n'ont pas étudiées chez ces patients.
Traitement combiné avec une sulfonylurée et/ou de l'insuline
Lorsque Jardiance Met est associée à une sulfonylurée et/ou à l'insuline, une réduction de la posologie du sulfamide hypoglycémiant et/ou de l'insuline pourrait être nécessaire pour diminuer le risque d'hypoglycémie (voir "Interactions" et "Effets indésirables" )
Oubli d'une dose
Lorsqu'une dose a été oubliée, le patient devrait la prendre dès qu'il s'en souvient. Néanmoins, il ne faut pas prendre deux doses au même moment; dans ce cas, il convient de ne pas prendre la dose oubliée.
Interruption temporaire en cas d'opérations
Si possible, Jardiance Met doit être arrêté au moins 3 jours avant une opération chirurgicale majeure ou des interventions associées à un jeûne prolongé. Jardiance Met peut être repris si le patient est cliniquement stable et consomme des aliments par voie orale (voir "Mises en garde et précautions" ).
Contre-indications-Hypersensibilité à l'empagliflozine et/ou la metformine ou à l'un des excipients selon la composition
-Toute forme d'acidose métabolique aiguë (p.ex. acidose lactique ou acidocétose diabétique)
-Coma et précoma diabétique
-Insuffisance rénale sévère (DFGe <30 ml/min/1,73 m2 ou clairance de la créatinine <30 ml/min)
-États critiques qui peuvent altérer la fonction rénale, tels que déshydratation (diarrhées, vomissements répétés), infection sévère p.ex. des voies urinaires, forte fièvre, état hypoxique sévère (choc, septicémie), administration intravasculaire de produit de contraste contenant de l'iode (voir section "Mises en garde et précautions" )
-Maladies (en particulier les pathologies aiguës ou l'aggravation d'une pathologie chronique) susceptibles de causer une hypoxie tissulaire, telles que, p. ex., insuffisance cardiaque décompensée, insuffisance respiratoire, infarctus du myocarde récent, septicémie ou choc. Dans ces situations, le risque de développement d'une acidose lactique est accru.
-Insuffisance hépatique
-Intoxication alcoolique aiguë, alcoolisme
Mises en garde et précautionsJardiance Met ne doit pas être utilisé chez les patients souffrant de diabète sucré de type 1.
Acidocétose diabétique
Des cas d'acidocétose diabétique (ACD), trouble grave du métabolisme mettant le pronostic vital en jeu et exigeant une hospitalisation immédiate, ont été rapportés pour des patients traités par empagliflozine, dont des cas à issue fatale. Dans quelques-uns des cas rapportés, la maladie s'est manifestée de manière atypique avec des taux de glycémies modérés inférieurs à 14 mmol/l (250 mg/dl).
Le risque d'acidocétose diabétique (ACD) doit être pris en compte en cas d'apparition de symptômes non spécifiques tels que nausées, vomissements, manque d'appétit, douleurs abdominales, soif excessive, difficultés respiratoires, confusion, épuisement inexpliqué ou fatigue chez les patients diabétiques traités par l'empagliflozine.
Si ces symptômes apparaissent, un test permettant de déceler la présence éventuelle de corps cétoniques doit être immédiatement effectué chez ces patients indépendamment du taux de glycémie. En cas de suspicion d'acidocétose, le traitement par Jardiance Met doit être arrêté, l'état du patient doit être examiné et un traitement immédiat doit être démarré. L'acidocétose diabétique peut durer plus longtemps chez certains patients après l'arrêt de Jardiance Met, c'est-à-dire qu'elle peut durer plus longtemps que prévu sur la base de la demi-vie plasmatique de l'empagliflozine. Une glycosurie prolongée a été observée, ainsi qu'une acidocétose diabétique persistante. L'excrétion urinaire de glucose persiste jusqu'à 3 jours après l'arrêt de Jardiance Met; cependant, il existe des rapports post-commercialisation d'acidocétose diabétique et de glucosurie qui durent plus de 6 jours et parfois jusqu'à 2 semaines après l'arrêt des inhibiteurs du SGLT2.
Un risque accru d'acidocétose peut exister lors de la prise de Jardiance Met chez les patients qui ont une nourriture très pauvre en hydrates de carbones (car l'association pourrait augmenter la production de corps cétoniques), chez les patients souffrant d'une pathologie aiguë, en cas de maladies du pancréas indiquant un manque d'insuline (p.ex. diabète sucré de type 1, pancréatite ou opération du pancréas dans l'anamnèse), en cas de réduction de la dose d'insuline (y compris défaillance de la pompe d'insuline), en cas d'abus d'alcool et de forte déshydratation ainsi que chez les patients qui ont déjà eu par le passé une acidocétose. Jardiance Met doit être utilisé avec prudence chez ces patients. La prudence est de rigueur en cas de réduction de la dose d'insuline [voir "Posologie/Mode d'emploi" ]. Dans les situations cliniques dont on sait qu'elles prédisposent à une acidocétose (p.ex. jeûne prolongé en raison d'une affection aiguë, d'une intervention ou d'une opération chirurgicale), une surveillance de l'acidocétose est indiquée et le traitement par Jardiance Met doit être temporairement interrompu. Dans ces situations, une surveillance des corps cétoniques doit également être envisagée, même si le traitement par Jardiance Met est interrompu. Le traitement par Jardiance Met peut être poursuivi si le patient est cliniquement stable et consomme des aliments par voie orale (voir "Posologie/Mode d'emploi" ).
Acidose lactique
L'acidose lactique est une complication métabolique très rare, mais grave. Les facteurs de risque comprennent le diabète mal contrôlé, la cétose, le jeûne prolongé, la consommation excessive d'alcool, les infections graves, la septicémie, l'insuffisance hépatique et toutes les situations associées à une hypoxie, par exemple l'insuffisance cardiaque décompensée, les affections cardiorespiratoires ou les infarctus aigus du myocarde. Il faut également être prudent lorsque l'on combine Jardiance Met avec des médicaments qui peuvent causer une acidose lactique, comme les inhibiteurs nucléosidiques de la transcriptase inverse.
Le risque d'une acidose lactique augmente avec le degré du dysfonctionnement rénal et l'âge du patient. En cas de traitement par Jardiance Met, la fonction rénale doit être contrôlée régulièrement. Une surveillance attentive est nécessaire, en particulier, chez les patients âgés. Une acidose lactique peut survenir en raison de l'accumulation de metformine. Dans la plupart des cas d'acidose lactique sous metformine connus à ce jour, les patients souffraient d'insuffisance rénale aiguë ou d'une dégradation aiguë de la fonction rénale. Il faut donc faire preuve d'une prudence particulière dans les situations où la fonction rénale peut se dégrader de façon aiguë, p.ex. en cas de déshydratation (diarrhée sévère, fièvre ou vomissements répétés), début d'un traitement avec des inhibiteurs de l'ECA et des antagonistes du récepteur de l'angiotensine II, des diurétiques, en particulier des diurétiques de l'anse ou des antirhumatismaux non stéroïdiens, y compris des inhibiteurs sélectifs de la cyclo-oxygénase 2 (COX-2). En cas de suspicion d'acidose lactique, le patient doit être hospitalisé immédiatement. Le lactate et la metformine sont éliminés le plus efficacement par hémodialyse (voir Surdosage). Il faut mettre les patients en garde contre une consommation excessive d'alcool, aiguë ou chronique, car l'alcool potentialise l'effet de la metformine sur le métabolisme du lactate.
Le médecin doit informer les patients et/ou leurs soignants du risque et des symptômes de l'acidose lactique. Il faut également demander aux patients de suspendre immédiatement la prise de Jardiance Met si ces symptômes évocateurs se manifestent et de consulter très rapidement un médecin. Le traitement par Jardiance Met ne doit pas être repris tant que la situation n'est pas clarifiée. Avant d'envisager la reprise du traitement par Jardiance Met, il convient d'évaluer le rapport bénéfice-risque individuel et d'examiner la fonction rénale.
Diagnostic
Les symptômes non spécifiques suivants peuvent être un signe d'acidose lactique: crampes musculaires, troubles gastro-intestinaux, tels que des douleurs abdominales, et une asthénie sévère. L'acidose lactique se caractérise par une dyspnée acidosique, des douleurs abdominales et une hypothermie suivies d'un coma. Le diagnostic est basé sur les analyses de laboratoire suivantes: diminution du pH sanguin (<7,35), concentration accrue de lactate plasmatique > 5 mmol/l et augmentation du déficit anionique et du rapport lactate/pyruvate.
Patients présentant une maladie mitochondriale connue ou présumée
Chez les patients présentant une maladie mitochondriale connue, telle que le syndrome d'encéphalomyopathie mitochondriale, acidose lactique et pseudo-épisodes vasculaires cérébraux (syndrome MELAS) et le diabète avec surdité de transmission maternelle (MIDD), la metformine n'est pas recommandée en raison du risque d'exacerbation de l'acidose lactique et de complications neurologiques pouvant entraîner une aggravation de la maladie.
En cas de signes et symptômes évocateurs du syndrome MELAS ou du MIDD après la prise de metformine, le traitement par Jardiance Met doit être interrompu immédiatement et un bilan diagnostique doit être réalisé rapidement.
Fasciite nécrosante du périnée (gangrène de Fournier)
Des cas de fasciite nécrosante du périnée (aussi appelée "gangrène de Fournier" ) ont été rapportés chez des patients de sexe masculin et féminin traités par des inhibiteurs du SGLT2, dont l'empagliflozine également. Il s'agit d'une infection nécrosante rare, mais grave et menaçant le pronostic vital. Parmi les conséquences graves comptaient hospitalisations, multiples opérations et décès. Les patients traités par Jardiance Met et rapportant des douleurs ou une sensibilité à la pression, des érythèmes ou une tuméfaction dans la région génitale ou périnéale, de la fièvre ou un malaise doivent être examinés à la recherche d'une fasciite nécrosante. En cas de suspicion de fasciite nécrosante, le traitement par Jardiance Met doit être arrêté et un traitement (entre autres par des antibiotiques à large spectre et, le cas échéant, par un débridement chirurgical) doit être instauré immédiatement.
Fonction rénale
En raison de son mécanisme d'action, une diminution de la fonction rénale entraîne une baisse d'efficacité de l'empagliflozine. Le DFGe doit être évalué avant le début du traitement et régulièrement par la suite (voir "Instructions spéciales pour la posologie" ). Jardiance Met n'est pas recommandé chez les patients avec un DFGe < 45 ml/min/1,73m2 et est contre-indiqué chez les patients avec un DFGe < 30 ml/min/1,73m2.
Des contrôles intensifs sont indiqués dans toutes les situations cliniques dans lesquelles une détérioration rapide de la fonction rénale est prévisible.
Une prudence particulière est de rigueur dans les cas où la fonction rénale pourrait se détériorer en raison de facteurs de prédisposition sous-jacents ou d'une éventuelle médication associée (p.ex. chez les patients âgés, en cas de déshydratation, au début d'un traitement par diurétiques, antihypertenseurs ou antirhumatismaux non stéroïdiens). Dans ces cas, il est également recommandé d'examiner la fonction rénale avant le début du traitement et à intervalles réguliers par la suite. Dans certains cas, l'interruption temporaire de Jardiance Met peut être envisagée.
Fonction cardiaque
Les patients souffrant d'insuffisance cardiaque ont un risque plus élevé d'hypoxie et d'insuffisance rénale. Chez les patients présentant une insuffisance cardiaque chronique stable, il est possible d'appliquer un traitement avec Jardiance Met, la fonction cardiaque et la fonction rénale devant alors être contrôlées régulièrement.
Chez les patients souffrant d'insuffisance cardiaque aiguë et décompensée, Jardiance Met est contre-indiqué (voir Contre-indications).
Patients âgés
Un risque accru d'hypovolémie existe éventuellement chez les patients âgés (≥75 ans). C'est pourquoi la prudence est de rigueur lors de la prescription de Jardiance Met dans ce groupe de patients (voir "Effets indésirables" ).
Hypovolémie
En raison de la diurèse osmotique, l'empagliflozine entraîne une légère baisse de la pression artérielle (la baisse systolique étant plus forte que la diastolique) et peut provoquer une hypotension orthostatique, ce qui peut entraîner des effets indésirables tels que vertiges, syncopes ou chutes. La prudence est donc de mise chez les patients avec hypotension orthostatique, les patients sous antihypertenseurs, les patients âgés ainsi que les patients avec affection cardiovasculaire et/ou cérébrovasculaire connue.
L'expérience montre que l'hématocrite augmente de 2 % environ.
En cas d'affections pouvant entraîner une perte hydrique (p.ex. les maladies gastro-intestinales), une surveillance attentive de l'état volémique et des électrolytes est recommandée chez les patients recevant de l'empagliflozine. L'interruption temporaire du traitement par Jardiance Met doit être envisagée jusqu'à ce que la perte hydrique soit corrigée.
Infections urinaires
Le traitement par empagliflozine accroît le risque d'infections urinaires. Elles concernent fréquemment les patients avec une anamnèse connue d'infections urinaires chroniques ou récidivantes et les femmes. Après la mise sur le marché, des cas d'infection des voies urinaires avec des complications incluant des pyélonéphrites et des urosepsis, ont été signalés chez les patients traités par l'empagliflozine. Le médecin traitant doit prêter attention aux signes possibles d'infection des voies urinaires et débuter un traitement immédiatement. Chez les patients présentant des infections urinaires avec complications (p.ex. pyélonéphrite ou urosepsis), une interruption temporaire du traitement doit être envisagée.
Amputations des membres inférieurs
Une augmentation du nombre de cas d'amputation des membres inférieurs (principalement d'un orteil) a été observée au cours d'études cliniques à long terme menées avec un autre inhibiteur du SGLT2. On ignore s'il s'agit d'un effet de classe. Comme pour tous les patients diabétiques, il est important de sensibiliser les patients sur l'importance des soins préventifs de routine pour les pieds.
Utilisation de produits de contraste iodés
L'administration intravasculaire de produits de contraste iodés pour les examens radiologiques peut entraîner une insuffisance rénale. Ceci peut provoquer une accumulation de metformine et une acidose lactique. Le traitement par Jardiance Met doit être arrêté avant ou au moment de l'examen d'imagerie et ne doit être repris qu'après un délai minimum de 48 heures, à condition que la fonction rénale ait été réévaluée et jugée stable.
Interventions chirurgicales
Jardiance Met contenant de la metformine, le traitement doit être arrêté 48 heures avant une intervention sous anesthésie générale, spinale ou péridurale. Le traitement ne doit être repris que 48 heures au plus tôt après l'intervention ou en cas de reprise de l'alimentation orale si la fonction rénale a été considérée comme suffisante (DGFe ≥60 ml/min/1,73 m2).
Evénements cérébrovasculaires
Dans le cadre de l'étude EMPA-REG OUTCOME, l'empagliflozine (groupes combinés traités par empagliflozine 10 mg et 25 mg) comparé au groupe placebo était associé à un risque supérieur d'AVC mortels/non mortels à une tendance non significative: HR 1,18 (IC à 95% 0,89; 1,56) (voir "Efficacité clinique" ). Un lien causal entre l'empagliflozine et l'AVC n'est pas prouvé. Il est néanmoins recommandé de faire preuve de prudence avec les patients affichant un risque élevé de survenue d'événements cérébrovasculaires.
Vitamine B12
La metformine peut diminuer les concentrations sériques de vitamine B12. Le risque de diminution des concentrations de vitamine B12 augmente simultanément à la hausse de la dose de metformine, la prolongation de la durée de traitement et chez les patients exposés à des facteurs de risque de carence en vitamine B12. Il est recommandé de contrôler les concentrations sériques de vitamine B12 à intervalles réguliers (par ex. tous les ans) ainsi qu'en cas de suspicion de carence en vitamine B12 (p.ex. en cas d'anémie ou de neuropathie).
Enfants et adolescents
L'étude pédiatrique DINAMO dans laquelle des enfants et des adolescents atteints de diabète sucré de type 2 ont été traités par empagliflozine associée à la metformine (voir rubrique "Efficacité clinique" ) suggère un profil de sécurité comparable chez les enfants à partir de 10 ans, les adolescents et les adultes atteints de diabète sucré de type 2. Les mises en garde et précautions mentionnées pour les adultes sont donc également valables pour la population pédiatrique.
Dans l'étude DINAMO, aucune différence pertinente entre le placebo et l'empagliflozine en termes de croissance ou de maturité sexuelle n'a été rapportée après 26 semaines de traitement. De même, des études cliniques contrôlées réalisées sur une période de 1 an n'ont montré aucun effet de la metformine sur la croissance ou la puberté. On ne dispose cependant pas de données concernant une durée de traitement plus longue. Par conséquent, un suivi attentif des paramètres de croissance chez les enfants, en particulier avant la puberté, est recommandé.
InteractionsGénérales
Chez les sujets sains, l'administration simultanée et répétée d'empagliflozine (50 mg une fois par jour) et de metformine (1000 mg deux fois par jour) ne modifie pas la pharmacocinétique de l'empagliflozine ou de la metformine.
Il n'y a pas d'études pharmacocinétiques sur les interactions avec Jardiance Met, mais des études avec chaque principe actif seul (à savoir l'empagliflozine et la metformine) ont été réalisées.
Empagliflozine
Interactions pharmacodynamiques
Diurétiques
L'empagliflozine peut majorer l'effet diurétique des thiazidiques et des diurétiques de l'anse et augmenter le risque de déshydratation et d'hypotension.
Insuline et sécrétagogues de l'insuline
L'insuline et les sécrétagogues de l'insuline, comme les sulfonylurées, peuvent augmenter le risque d'hypoglycémie. Par conséquent, une réduction de la dose d'insuline ou du sécrétagogue de l'insuline peut être nécessaire pour diminuer le risque d'hypoglycémie lorsqu'ils sont utilisés en association avec l'empagliflozine.
Interactions pharmacocinétiques
Effets des autres médicaments sur l'empagliflozine
Les données in vitro suggèrent que la voie primaire du métabolisme de l'empagliflozine chez l'homme est la glucuronidation par les uridine-5'-diphosphoglucuronosyltransférases UGT1A3, UGT1A8, UGT1A9 et UGT2B7. L'empagliflozine est un substrat des transporteurs humains OAT3, OATP1B1 et OATP1B3, mais pas d'OAT1 et OCT2. L'empagliflozine est un substrat de la glycoprotéine P (P-gp) et de la protéine de résistance du cancer du sein (BCRP).
L'administration concomitante d'empagliflozine et de probénécide, un inhibiteur des enzymes UGT et de l'OAT3, a entraîné une augmentation de 26 % du pic de concentration plasmatique d'empagliflozine (Cmax) et une augmentation de 54 % de l'aire sous la courbe des concentrations en fonction du temps (ASC).
Ces variations n'ont pas été considérées comme cliniquement significatives.
L'effet de l'induction des UGT sur l'empagliflozine n'a pas été étudié.
Un traitement concomitant par des inducteurs connus des enzymes UGT doit être évité en raison du risque potentiel d'une diminution de l'efficacité.
Une étude d'interaction avec le gemfibrozil, un inhibiteur in vitro des transporteurs OAT3 et OATP1B1/1B3, a montré une augmentation de 15 % de la Cmax de l'empagliflozine et de 59 % de l'ASC en cas d'administration concomitante. Ces variations n'ont pas été considérées comme cliniquement significatives.
L'inhibition des transporteurs OATP1B1/1B3 par l'utilisation concomitante de rifampicine a entraîné une augmentation de 75 % de la Cmax de l'empagliflozine ainsi qu'une augmentation de 35 % de l'ASC.
Ces variations n'ont pas été considérées comme cliniquement significatives.
L'exposition à l'empagliflozine était similaire avec ou sans administration concomitante de vérapamil, un inhibiteur de la P-gp; ceci indique que l'inhibition de la P-gp n'a pas d'effet cliniquement significatif sur l'empagliflozine.
Les études d'interaction menées chez des volontaires sains suggèrent que la pharmacocinétique de l'empagliflozine n'est pas influencée par l'administration concomitante de metformine, de glimépiride (dose unique), de vérapamil, de ramipril, de simvastatine, de torasémide ou d'hydrochlorothiazide.
Effets de l'empagliflozine sur les autres médicaments
D'après les études in vitro, l'empagliflozine n'inhibe pas, n'inactive pas et n'induit pas les isoformes du CYP450. L'UGT1A1 n'est pas inhibée par l'empagliflozine.
Par conséquent, des interactions médicamenteuses, impliquant les principales isoformes du CYP450 ou l'UGT1A1, entre l'empagliflozine et des substrats co-administrés de ces enzymes, sont considérées comme peu probables. Le potentiel d'inhibition d'UGT2B7 de l'empagliflozine n'a pas été étudié.
À des doses thérapeutiques, l'empagliflozine n'inhibe pas la P-gp. Sur la base des études in vitro, il est considéré comme peu probable que l'empagliflozine entraîne des interactions avec des médicaments substrats de la P-gp. L'administration concomitante d'empagliflozine et de digoxine, un substrat de la P-gp, a entraîné une augmentation de 6 % de l'ASC et une augmentation de 15 % de la Cmax de la digoxine. Il convient donc de surveiller les patients sous digoxine.
À des concentrations cliniquement significatives, l'empagliflozine n'inhibe pas les transporteurs humains OAT3, OATP1B1 et OATP1B3 in vitro. C'est pourquoi les interactions médicamenteuses avec des substrats de ces transporteurs sont considérées comme peu probables.
Les études d'interaction menées chez des volontaires sains suggèrent que l'empagliflozine n'a pas d'influence cliniquement significative sur la pharmacocinétique de la metformine, du glimépiride, de la simvastatine, de la warfarine, du ramipril, de l'hydrochlorothiazide, de la torasémide et des contraceptifs oraux.
Aucune donnée n'est disponible pour l'acénocoumarol et la phenprocoumone.
Les inhibiteurs du SGLT2, y compris l'empagliflozine, peuvent augmenter l'excrétion rénale du lithium et entraîner une diminution des concentrations sanguines de lithium. La concentration sérique de lithium doit être contrôlée plus souvent après l'instauration du traitement par l'empagliflozine et en cas de modification de la posologie. Pour surveiller la concentration sérique du lithium, le patient doit être adressé au médecin ayant prescrit le lithium.
Metformine
Lors de l'administration simultanée des médicaments indiqués ci-dessous et de metformine, ainsi que de l'arrêt de ces médicaments au cours du traitement par la metformine, la glycémie doit être contrôlée étroitement. Les patients doivent être informés en conséquence. Si nécessaire, la posologie du traitement antidiabétique doit être ajustée pendant la durée de la thérapie concomitante.
Interactions influençant l'effet de la metformine
Diminution de l'effet hypoglycémiant
Les glucocorticoïdes (voie systémique et locale), les β2-sympathomimétiques, les diurétiques, les phénothiazines (par ex. chlorpromazine), les hormones thyroïdiennes, les estrogènes, les contraceptifs oraux, les hormones de substitution, la phénytoïne, l'acide nicotinique, les antagonistes calciques, l'isoniazide et le tétracosactide peuvent augmenter la glycémie.
Renforcement de l'effet hypoglycémiant
Le furosémide augmente la concentration plasmatique de la metformine (Cmax de 22 %, ASC de 15 %) sans modification significative de la clairance rénale.
La nifédipine augmente la concentration plasmatique de la metformine (Cmax de 20 %, ASC de 9-20 %) par l'augmentation de l'absorption de metformine.
La cimétidine augmente la Cmax de la metformine de 60 % et l'ASC de 40 %. La demi-vie d'élimination de la metformine n'est pas influencée. D'autres substances (amiloride, digoxine, morphine, procaïnamide, quinidine, quinine, ranitidine, triamtérène, triméthoprime ou vancomycine), éliminées par une sécrétion rénale tubulaire active, sont susceptibles d'entraîner une interaction avec la metformine. Les patients traités par ces médicaments doivent donc être surveillés étroitement lors du traitement par la metformine.
Les inhibiteurs de l'ECA peuvent aussi abaisser la glycémie.
La glycémie peut également être diminuée par des bêtabloquants; les bêtabloquants cardio-sélectifs (β1-sélectifs) ont nettement moins tendance à induire ces interactions que les non cardio-sélectifs.
L'administration concomitante d'inhibiteurs de la MAO et d'antidiabétiques oraux peut améliorer la tolérance au glucose et renforcer l'effet hypoglycémiant.
La consommation simultanée d'alcool peut renforcer l'effet hypoglycémiant de la metformine jusqu'au coma hypoglycémique.
Renforcement ou diminution de l'effet hypoglycémiant de la metformine
Les antagonistes H2, la clonidine et la réserpine peuvent renforcer ou atténuer l'effet de la metformine. Des troubles de la glycémie (y compris hyperglycémie ou hypoglycémie) ont été observés lors de l'administration simultanée de quinolones et de metformine.
Interactions pouvant augmenter les effets indésirables de la metformine
Diurétiques: une acidose lactique peut survenir en raison d'un trouble de la fonction rénale lié à la prise de diurétiques (particulièrement de l'anse). En outre, les diurétiques peuvent exercer un effet hyperglycémiant.
Produits de contraste iodés: pour les interactions avec les produits de contraste iodés et le risque d'une acidose lactique, voir la rubrique "Mises en garde et précautions" .
L'administration concomitante de metformine avec:
des substrats / inhibiteurs des OCT1, comme le vérapamil, peut réduire l'efficacité de la metformine.
des inducteurs des OCT1, comme la rifampicine, peut augmenter l'absorption gastro-intestinale et l'efficacité de la metformine.
des substrats / inhibiteurs des OCT2, comme la cimétidine, le dolutégravir, le crizotinib, l'olaparib, le daclatasvir, le vandétanib, peut diminuer l'élimination rénale et induire ainsi une augmentation de la concentration plasmatique.
La prudence est donc recommandée, en particulier chez les patients insuffisants rénaux, lorsque ces médicaments sont administrés concomitamment à la metformine car la concentration plasmatique de la metformine peut augmenter. Si nécessaire, et puisque les inhibiteurs/inducteurs des OCT peuvent entraîner une modification de l'efficacité de la metformine, un ajustement de la dose de metformine peut être envisagé.
Alcool: le risque d'acidose lactique sous metformine est majoré par une intoxication alcoolique aiguë, particulièrement en cas de jeûne antérieur ou en présence de dénutrition ou d'insuffisance hépatique. La consommation d'alcool ou de médicaments contenant de l'alcool doit être évitée.
Interactions influençant l'effet d'autres substances
La metformine abaisse la concentration plasmatique du furosémide (Cmax de 33 %, ASC de 12 %) dont la demi-vie terminale est réduite de 32 % sans modification de sa clairance rénale.
L'effet du phenprocoumone peut être diminué, car son élimination est accélérée par la metformine.
Des études d'interactions avec le glibenclamide, la nifédipine, l'ibuprofène ou le propranolol n'ont révélé aucun effet d'importance clinique sur les paramètres pharmacocinétiques de ces substances.
Autres interactions
Sous l'influence de substances à action sympatholytique (par ex. bêtabloquants, clonidine, guanéthidine, réserpine), la perception des signes avant-coureurs d'une hypoglycémie peut être masquée.
Interférence avec le dosage du 1,5-anhydroglucitol (1,5-AG)
Une surveillance de la glycémie au moyen du dosage du 1,5-AG n'est pas recommandée, car les taux de 1,5-AG ne permettent pas une mesure fiable du contrôle glycémique chez les patients traités par des inhibiteurs du SGLT2. D'autres méthodes devraient être utilisées pour surveiller la glycémie.
Enfants et adolescents
Des études des interactions ont été menées exclusivement chez des adultes.
Grossesse, allaitementGrossesse
Un diabète insuffisamment contrôlé pendant la grossesse (dû à la grossesse ou préexistant) est associé à une élévation du risque de malformations congénitales et de la mortalité périnatale.
Les données concernant l'utilisation de Jardiance Met ou de l'un de ses composants chez la femme enceinte sont limitées.
Les expérimentations animales réalisées avec l'empagliflozine n'ont pas révélé de toxicité de reproduction.
Les expérimentations animales réalisées avec l'association empagliflozine plus metformine ou avec la metformine en monothérapie n'ont mis en évidence un effet tératogène qu'à des doses élevées de metformine (voir "Données précliniques" ).
Jardiance Met ne doit pas être administrée pendant la grossesse sauf en cas d'absolue nécessité.
La glycémie doit être amenée à des valeurs aussi normales que possible avec l'insuline afin de réduire le risque de malformations et de complications supplémentaires pour l'enfant.
Allaitement
La metformine est excrétée dans le lait maternel humain en faible quantité. Elle est contre-indiquée pendant l'allaitement.
On ignore si l'empagliflozine passe dans le lait maternel. Les données disponibles provenant des expérimentations animales ont montré que l'empagliflozine passe dans le lait. Les expérimentations animales ont montré des effets indésirables sur le développement post-natal (voir "Données précliniques" ).
Un risque pour le nouveau-né/le nourrisson ne peut être exclu. C'est pourquoi l'association Jardiance Met contenant de la metformine est contre-indiquée pendant l'allaitement ou l'allaitement doit être interrompu pendant le traitement avec Jardiance Met.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée. Les patients doivent toutefois être prévenus du risque d'hypoglycémie lorsque Jardiance Met est utilisé comme traitement d'appoint à l'insuline et/ou une sulfonylurée. Ils doivent aussi être prévenus du risque majoré d'effets indésirables liés à la réduction du volume intravasculaire, p.ex. des vertiges (voir "Mises en garde et précautions" et "Effets indésirables" ).
Effets indésirablesDans des études de sécurité, un total de 12245 patients avec un diabète sucré de type 2 ont été traités par empagliflozine, dont 8199 patients recevant de l'empagliflozine plus de la metformine seule ou associée à une sulfonylurée, à la pioglitazone, aux inhibiteurs du DPP4 ou à l'insuline.
Le profil général de sécurité de l'empagliflozine plus metformine chez les patients qui ont pris part à l'étude EMPA-REG-OUTCOME était comparable aux profils de sécurité précédemment connus des deux substances individuelles.
Plusieurs études en double aveugle contrôlées contre placebo d'une durée de 18 à 24 semaines ont inclus 3456 patients, dont 1271 ont été traités par l'empagliflozine 10 mg plus metformine et 1259 par l'empagliflozine 25 mg plus metformine.
L'effet indésirable le plus fréquemment signalé dans le cadre des études cliniques était une hypoglycémie, en fonction du traitement de fond utilisé dans les différentes études (voir la description des effets indésirables spéciaux).
Dans le cadre des études cliniques réalisées avec l'empagliflozine plus metformine aucun autre effet indésirable n'a été observé en comparaison avec chaque composant administré seul.
Les fréquences des effets indésirables sont définies de la façon suivante: "très fréquent" (≥1/10), "fréquent" (<1/10, ≥1/100), "occasionnel" (<1/100, ≥1/1000), "rare" (<1/1000, ≥1/10 000) ou "très rare" (<1/10 000).
Les effets indésirables suivants ont été rapportés chez les patients traités par empagliflozine plus metformine dans des études en double aveugle contrôlées contre placebo d'une durée maximale de 24 semaines (présentés par classe d'organe et en utilisant les termes préférés selon la classification MedDRA)
Infections et infestations
Fréquent: Candidose vaginale, vulvovaginite, balanite et autres infections des voies génitales1,2, Infections des voies urinaires1,2 (incluant des pyélonéphrites et des urosepticémies)6.
Fréquence inconnue: Fasciite nécrosante du périnée (gangrène de Fournier)2,6.
Troubles du métabolisme et de la nutrition
Très fréquent: Hypoglycémie (lors de l'association à une sulfonylurée ou à l'insuline)1.
Fréquent: Diminution des concentrations/carence en vitamine B12³.
Très rare: Acidose lactique³.
Affections du système nerveux
Fréquent: Troubles du goût (goût métallique)3.
Affections vasculaires
Occasionnel: Hypovolémie1,2.
Affections gastro-intestinales4
Très fréquent: Troubles gastro-intestinaux (5 à 15 %) tels que nausées3, vomissements3, diarrhées3, douleurs abdominales3, perte d'appétit3.
Fréquent: Constipation.
Affections hépatobiliaires
Très rare: résultats pathologiques des paramètres hépatiques anomalies des tests hépatiques, p.ex. augmentation des transaminases3 ou hépatite3 (réversible à l'arrêt de la metformine).
Affections de la peau et du tissu sous-cutané
Fréquent: Réactions cutanées allergiques (par ex. érythème3, éruption cutanée6, urticaire3,6, prurit3,2.
Cas isolés: Angioœdème2,6.
Affections du rein et des voies urinaires
Fréquent: Augmentation des mictions1,2.
Occasionnel: Dysurie2.
Très rare: Néphrite tubulo-interstitielle.
Troubles généraux
Fréquent: Soif.
Investigations
Fréquent: Augmentation des lipides sériques5, augmentation de l'hématocrite5.
Occasionnel: Baisse du débit de filtration glomérulaire1, augmentation de la créatinine sanguine1.
1 Informations complémentaires voir "Descriptions des effets indésirables spéciaux" .
2 Effets indésirables qui sont survenus lors d'un traitement par l'empagliflozine en monothérapie
3 Effets indésirables identifiés sur la base du résumé des caractéristiques de produit de l'UE pour la metformine
4 Les symptômes gastro-intestinaux tels que la nausée, les vomissements, la diarrhée, les douleurs abdominales et la perte d'appétit surviennent généralement au début du traitement et disparaissent spontanément dans la plupart des cas.
5 Voir "Efficacité clinique" pour plus d'informations
6 provenant de l'expérience acquise depuis la commercialisation
Dans le cadre de la surveillance après l'introduction sur le marché, des cas d'acidocétose diabétique ont été observés chez des patients traités avec des inhibiteurs du SGLT2, tels que l'empagliflozine (voir "Mises en garde et précautions" )
Descriptions des effets indésirables spéciaux
Les fréquences mentionnées des effets indésirables ci-dessous sont indiquées indépendamment d'une relation causale.
Hypoglycémie
La fréquence des hypoglycémies dépendait du traitement de fond dans les différentes études.
Hypoglycémie mineure
La fréquence des patients avec une hypoglycémie mineure a été similaire dans les groupes empagliflozine plus metformine et placebo plus metformine en monothérapie ou comme traitement d'appoint de la pioglitazone. Une augmentation de la fréquence a été observée lorsque l'empagliflozine était associée à un traitement d'appoint par une sulfonylurée (empagliflozine plus metformine 10 mg: 16,1 %, empagliflozine plus metformine 25 mg: 11,5 %, placebo plus metformine: 8,4 %). Comme traitement d'appoint à l'insuline (empagliflozine plus metformine 10 mg: 31,3 %, empagliflozine plus metformine 25 mg: 36,2 %, placebo plus metformine: 34,7 %) l'empagliflozine n'a pas montré d'augmentation du risque d'hypoglycémie par rapport au placebo. Comme traitement d'appoint à l'insuline plus une sulfonylurée, des hypoglycémies ont été rapportées chez 16,2 % des patients (empagliflozine plus metformine 10 mg), 32,8 % des patients (empagliflozine plus metformine 25 mg) et chez 13,2 % des patients (placebo plus metformine).
Hypoglycémie majeure (hypoglycémie exigeant un traitement)
Le nombre de patients présentant des événements hypoglycémiques majeurs était faible (<1 %) et similaire dans les groupes empagliflozine plus metformine et placebo plus metformine.
Candidose vaginale, vulvovaginite, balanite et autres infections des voies génitales
Des candidoses vaginales, vulvovaginites, balanites et autres infections génitales étaient plus fréquentes dans le groupe sous empagliflozine 10 mg plus metformine (4,0 %) et sous empagliflozine 25 mg plus metformine (3,9 %) par rapport au placebo plus metformine (1,3 %); comparé au groupe placebo, les femmes sous empagliflozine étaient plus souvent atteintes et la différence de fréquence était moins prononcée chez les hommes. Les infections de l'appareil génital étaient d'intensité légère ou modérée, des infections graves ne se sont pas produites.
Des cas de phimosis/phimosis acquis ont été rapportés concomitamment à des infections génitales.
Augmentation des mictions
Ainsi qu'attendu au vu du mécanisme d'action, une augmentation des mictions (comprenant les termes génériques [PT]: pollakiurie, polyurie et nycturie) a été observée plus fréquemment chez les patients traités par l'empagliflozine 10 mg plus metformine (3,0 %) et l'empagliflozine 25 mg plus metformine (2,9 %) que sous placebo plus metformine (1,4 %). L'intensité de cet effet était principalement légère à modérée. La fréquence de la nycturie rapportée était similaire pour le placebo et l'empagliflozine (tous deux associés à la metformine comme traitement de fond) (<1 %).
Hypovolémie
La fréquence globale des hypovolémies (comprenant les termes génériques de diminution de la pression artérielle [mesure ambulatoire], diminution de la pression artérielle systolique, déshydratation, hypotension, hypovolémie, hypotension orthostatique et syncope) a été faible et similaire à celle sous placebo (empagliflozine 10 mg plus metformine (0,6 %), empagliflozine 25 mg plus metformine (0,3 %) et placebo plus metformine (0,1 %). L'action de l'empagliflozine sur l'élimination du glucose avec l'urine est associée à une diurèse osmotique, ce qui pourrait influencer le statut d'hydratation des patients âgés de 75 ans et plus.
Augmentation de la créatinine sanguine et baisse du débit de filtration glomérulaire
La fréquence globale des patients présentant une augmentation de la créatinine sanguine et une baisse du débit de filtration glomérulaire a été comparable sous empagliflozine et placebo en thérapie adjuvante à la metformine (augmentation de la créatinine sanguine: empagliflozine 10 mg 0,5 %, empagliflozine 25 mg 0,1 %, placebo 0,4 %; baisse du débit de filtration glomérulaire: empagliflozine 10 mg 0,1 %, empagliflozine 25 mg 0 %, placebo 0,2 %).
Dans ces études en double aveugle contrôlées par placebo menées sur une période allant jusqu'à 24 semaines, des augmentations initiales temporaires de la créatinine sanguine (variation moyenne par rapport à la valeur initiale à la semaine 12: empagliflozine 10 mg 0,02 mg/dl, empagliflozine 25 mg 0,02 mg/dl) ainsi que des baisses initiales temporaires du débit de filtration glomérulaire estimé (variation moyenne par rapport à la valeur initiale à la semaine 12: empagliflozine 10 mg -1,46 ml/min/1,73 m2, empagliflozine 25 mg -2,05 ml/min/1,73 m2) ont été observées. Ces variations étaient généralement réversibles en cas de poursuite du traitement ou après arrêt de la médication.
Enfants et adolescents
L'étude DINAMO a inclus 157 enfants à partir de 10 ans atteints de diabète sucré de type 2: 52 patients ont été traités par empagliflozine, 52 patients par linagliptine et 53 patients ont reçu un placebo (voir rubrique relative aux études cliniques). Plus de 90% des participants à l'étude ont reçu en association un traitement par metformine.
Pendant la phase contrôlée contre placebo, l'effet médicamenteux indésirable le plus fréquent a été l'hypoglycémie (empagliflozine 10 mg et 25 mg, données poolées: 23,1%, placebo: 9,4%).
Aucun de ces événements n'a été sévère ou n'a nécessité un traitement.
Au total, le profil de sécurité chez les enfants a été comparable au profil de sécurité des patients adultes atteints de diabète sucré de type 2.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageSignes et symptômes
Dans le cadre d'études cliniques contrôlées avec des volontaires sains, des doses uniques jusqu'à 800 mg d'empagliflozine ont été administrées (correspondant à 32 fois la dose journalière maximale recommandée).
Aucune hypoglycémie n'a été observée avec des doses de chlorhydrate de metformine allant jusqu'à 85 g; en revanche, une acidose lactique a été constatée dans ces conditions. En cas de surdosage important ou de risques associés, le chlorhydrate de metformine peut induire une acidose lactique. L'acidose lactique est une urgence médicale qui requiert l'hospitalisation du patient.
Traitement
En cas de surdosage, un traitement approprié doit être initié en fonction de l'état clinique du patient. L'hémodialyse est la méthode la plus efficace pour éliminer le lactate et le chlorhydrate de metformine. L'élimination de l'empagliflozine par hémodialyse n'a pas été étudiée.
Propriétés/EffetsCode ATC
A10BD20
Classe pharmacothérapeutique:
Association avec des antidiabétiques oraux.
Mécanisme d'action / Pharmacodynamique
L'empagliflozine est un inhibiteur compétitif sélectif, réversible et très puissant (CI50 de 1,3 nmol) du cotransporteur du SGLT2. Il montre une sélectivité 5000 fois supérieure pour SGLT2 par rapport au SGLT1 humain responsable de l'absorption intestinale du glucose (CI50: 6278 nmol).
Un autre effet inhibiteur des transporteurs du glucose GLUT, qui sont responsables du transport du glucose dans les différents tissus, n'a pu être montré.
SGLT2 est principalement exprimé dans les reins. En tant que transporteur prédominant, il est responsable de la réabsorption du glucose du filtrat glomérulaire vers la circulation générale.
L'empagliflozine réduit la réabsorption rénale du glucose d'une manière indépendante de l'insuline. La quantité de glucose éliminée par le rein via ce mécanisme dépend de la glycémie et du taux de filtration glomérulaire.
L'augmentation de l'excrétion rénale du glucose entraîne une diurèse osmotique et, par cet effet diurétique, une baisse de la pression artérielle (4-5 mmHg en moyenne pour la pression systolique et 1-2 mmHg pour la diastolique) ainsi qu'une augmentation de l'hématocrite (environ 2-3 %). L'empagliflozine a également un effet uricosurique et réduit ainsi le taux plasmatique d'urée (environ 50 µmol/L). L'élimination rénale du glucose augmente le risque d'infections urogénitales, particulièrement chez les femmes et les personnes âgées.
L'élimination rénale du glucose après la prise de 10 mg d'empagliflozine est de 64 g par jour (correspondant à environ 256 kcal). Après la prise de 25 mg d'empagliflozine, l'élimination rénale du glucose est de 78 g par jour (correspondant à environ 312 kcal).
Chez les patients avec diabète de type 2, l'élimination rénale du glucose dans l'urine augmente après la première dose d'empagliflozine pour se maintenir au même niveau pendant tout l'intervalle de dosage de 24 heures.
L'augmentation de la glycosurie a mené à une réduction de la concentration plasmatique de glucose chez les patients avec un diabète de type 2.
L'effet hypoglycémiant de l'empagliflozine est indépendant de la fonction des cellules bêta et des voies d'activation de l'insuline.
La metformine est un biguanide aux effets antihyperglycémiants sur l'hyperglycémie basale et postprandiale. La metformine ne stimule pas la sécrétion d'insuline et n'entraîne pas d'hypoglycémie. La metformine réduit l'hyperinsulinémie basale et, en association avec l'insuline, en diminue les besoins. La metformine exerce ses effets antihyperglycémiants via plusieurs mécanismes:
La metformine diminue la production de glucose hépatique.
La metformine facilite l'absorption et l'assimilation périphérique du glucose, en partie en augmentant l'action de l'insuline.
La metformine modifie le métabolisme du glucose dans l'intestin: l'absorption par la circulation sanguine est augmentée et celle par l'alimentation est diminuée. Parmi les autres mécanismes attribués à l'intestin, figurent une libération accrue de glucagon-like peptide-1 (GLP-1) et une diminution de la résorption des acides biliaires. La metformine modifie le microbiome intestinal.
La metformine peut améliorer le profil lipidique chez les personnes souffrant d'hyperlipidémie.
Dans le cadre des études cliniques, l'utilisation de la metformine était associée à un poids corporel stable ou à une perte de poids modérée.
La metformine est un activateur de la protéine kinase activée par l'adénosine monophosphate (AMPK) et augmente la capacité de transport de tous les types de transporteurs transmembranaires du glucose (GLUT).
Efficacité clinique
Au total, 10224 patients présentant un diabète sucré de type 2 ont été traités pendant 24 semaines au moins dans le cadre de 9 études en double-aveugle contrôlées contre placebo ou contre comparateur actif; parmi ceux-ci 2947 patients ont reçu de l'empagliflozine 10 mg et 3703 de l'empagliflozine 25 mg en appoint du traitement par la metformine. Au total, 2978 de ces patients ont été traités par la metformine plus l'insuline, dont 988 avec l'empagliflozine 10 mg et 990 avec l'empagliflozine 25 mg comme traitement d'appoint.
Le traitement par empagliflozine en association avec la metformine et avec ou sans traitement de fond (sulfonylurée, inhibiteurs de la DPP4 ou l'insuline), a entraîné des améliorations cliniquement significatives de l'HbA1c, de la glycémie à jeun (GAJ), du poids corporel ainsi que de la pression artérielle systolique et diastolique.
Chez les patients à partir de 75 ans de faibles réductions numériques de l'HbA1c ont été observées. Des valeurs d'HbA1c plus élevées par rapport aux valeurs initiales sont associées à une réduction plus importante de l'HbA1c.
Association thérapeutique
Empagliflozine comme traitement d'appoint à la metformine et à une sulfonylurée
Par rapport au placebo, l'empagliflozine comme traitement d'appoint à la metformine ou à la metformine avec une sulfonylurée a entraîné une réduction statistiquement significative (p<0,0001) de l'HbA1c et du poids corporel (Tableau 2). De plus, elle a entraîné une réduction cliniquement significative de la glycémie à jeun et de la pression artérielle systolique et diastolique par rapport au placebo.
Dans la période d'extension menée en double aveugle et avec contrôle placebo de ces études, les réductions de l'HbA1c, du poids corporel et de la pression artérielle ont été maintenues jusqu'à la semaine 52.
Tableau 2: Résultats d'une étude de 24 semaines (LOCF) contrôlée contre placebo avec l'empagliflozine comme traitement d'appoint à la metformine ou à la metformine et une sulfonylurée (Full Analysis Set)
Empagliflozinecomme traitement Placebo Empagliflozine10 mg Empagliflozine25 mg
d'appoint à la metformine
N 207 217 213
HbA1c (%)
Valeur initiale (moyenne) 7,90 7,94 7,86
Variation par rapport à la valeur -0,13 -0,70 -0,77
initiale1
Différence par rapport au -0,57* (-0,72, -0,64* (-0,79,
placebo1(IC 97,5%) -0,42) -0,48)
N 184 199 191
Patients (%) atteignant une 12,5 37,7 38,7
valeur d'HbA1c <7% (valeur
initiale d'HbA1c ≥7%)2
N 207 217 213
Poids corporel (kg)
Valeur initiale (moyenne) 79,73 81,59 82,21
Variation par rapport à la valeur -0,45 -2,08 -2,46
initiale1
Différence par rapport au -1,63* (-2,17, -2,01* (-2,56,
placebo1(IC 97,5%) -1,08) -1,46)
N 207 217 213
Patients (%) avec une perte de 4,8 21,2 23,0
poids >5%
N 207 217 213
PAS (mmHg)2
Valeur initiale (moyenne) 128,6 129,6 130,0
Variation par rapport à la valeur -0,4 -4,5 -5,2
initiale1
Différence par rapport au -4,1* (-6,2, -2,1) -4.8* (-6.9, -2.7)
placebo1(IC 95%)
Empagliflozinecomme Placebo Empagliflozine10 mg Empagliflozine25 mg
traitement d'appoint à la
metformine et une
sulfonylurée
N 225 225 216
HbA1c (%)
Valeur initiale (moyenne) 8,15 8,07 8,10
Variation par rapport à la -0,17 -0,82 -0,77
valeur initiale1
Différence par rapport au -0,64* (-0,79, -0,49) -0,59* (-0,74, -0,44)
placebo1(IC 97,5%)
N 216 209 202
Patients (%) atteignant une 9,3 26,3 32,2
valeur d'HbA1c <7% (valeur
initiale d'HbA1c ≥7%)2
N 224 225 215
GAJ (mg/dl) [mmol/l]
Valeur initiale (moyenne) 151,7 [8,42] 151,0 [8,38] 156,5 [8,68]
Variation par rapport à la 5,5 [0,31] -23,3 [-1,29] -23,3 [-1,29]
valeur initiale1
Différence par rapport au -28,8* (-34,3, -23,4) -28,8* (-34,3, -23,3)
placebo1(IC 95%) [-1,60 (-1,90, -1,30)] [-1,60 (-1,90, -1,29)]
N 225 225 216
Poids corporel (kg)
Valeur initiale (moyenne) 76,23 77,08 77,50
Variation par rapport à la -0,39 -2,16 -2,39
valeur initiale1
Différence par rapport au -1,76* (-2,25, -1,28) -1,99* (-2,48, -1,50)
placebo1(IC 97,5%)
N 225 225 216
Patients (%) avec une perte 5,8 27,6 23,6
de poids >5%
N 225 225 216
PAS (mmHg)2
Valeur initiale (moyenne) 128,8 128,7 129,3
Variation par rapport à la -1,4 -4,1 -3,5
valeur initiale1
Différence par rapport au -2,7 (-4,6, -0,8) -2,1 (-4,0, -0,2)
placebo1(IC 95%)
1 Moyenne ajustée par rapport à la valeur initiale
2 Non évalué en termes de signification statistique du fait de la procédure d'analyse de confirmation séquentielle
* p<0,0001
Traitement d'appoint à l'insuline
Empagliflozine comme traitement d'appoint à l'insuline basale
L'efficacité et la tolérance de l'empagliflozine comme traitement d'appoint à l'insuline basale avec ou sans metformine et/ou une sulfonylurée ont été évaluées au cours d'une étude en double aveugle avec contrôle placebo d'une durée de 78 semaines. Pendant les 18 premières semaines, la dose d'insuline a été maintenue à dose constante, mais elle a été ajustée au cours des 60 semaines suivantes pour obtenir une glycémie à jeun <110 mg/dl.
Jusqu'à la semaine 18, l'empagliflozine a entraîné une amélioration statistiquement significative de l'HbA1c (Tableau 3).
Après la semaine 78, le traitement par empagliflozine a entraîné une réduction statistiquement significative de l'HbA1c ainsi qu'une épargne insulinique par rapport au placebo. De plus, l'empagliflozine a également entraîné une réduction de la glycémie à jeun, du poids corporel et de la pression artérielle.
Tableau 3: Résultats d'une étude avec contrôle placebo sur l'empagliflozine comme traitement d'appoint à l'insuline basale avec ou sans metformine ou une sulfonylurée (Full Analysis Set – Compléter) après 18 ou 78 semaines (LOCF)
Placebo Empagliflozine10 mg Empagliflozine25 mg
N 125 132 117
HbA1c (%) à la semaine 18
Valeur initiale (moyenne) 8,10 8,26 8,34
Variation par rapport à la -0,01 -0,57 -0,71
valeur initiale1
Différence par rapport au -0,56* (-0,78, -0,33) -0,70* (-0,93, -0,47)
placebo1(IC 97,5 %)
N 112 127 110
HbA1c (%) à la semaine 78
Valeur initiale (moyenne) 8,09 8,27 8,29
Variation par rapport à la -0,02 -0,48 -0,64
valeur initiale1
Différence par rapport au -0,46* (-0,73, -0,19) -0,62* (-0,90, -0,34)
placebo1(IC 97,5 %)
N 112 127 110
Dose d'insuline basale
(UI/jour) à la semaine 78
Valeur initiale (moyenne) 47,84 45,13 48,43
Variation par rapport à la 5,45 -1,21 -0,47
valeur initiale1
Différence par rapport au -6,66** (-11,56, -5,92** (-11,00,
placebo1(IC 97,5 %) -1,77) -0,85)
1 Moyenne ajustée par rapport à la valeur initiale
* p <0,0001
** p <0,01
Empagliflozine comme traitement d'appoint à l'insuline injectée plusieurs fois par jour
L'efficacité et la tolérance de l'empagliflozine comme traitement d'appoint à l'insuline administrée plusieurs fois par jour avec ou sans traitement associé par metformine ont été évaluées au cours d'une étude en double aveugle avec contrôle placebo d'une durée de 52 semaines. Pendant les 18 premières semaines et les 12 dernières semaines, la dose d'insuline a été maintenue constante, mais elle a été ajustée entre les semaines 19 et 40 pour obtenir des glycémies préprandiales <100 mg/dl (5,5 mmol/l), et des glycémies postprandiales <140 mg/dl (7,8 mmol/l).
Jusqu'à la semaine 18, l'empagliflozine a entraîné une amélioration statistiquement significative de l'HbA1c par rapport au placebo (Tableau 4).
Après la semaine 52, le traitement par empagliflozine a entraîné une réduction statistiquement significative de l'HbA1c ainsi qu'une épargne insulinique par rapport au placebo. De plus, l'empagliflozine a également entraîné une réduction de la GAJ et du poids corporel.
Tableau 4: Résultats d'efficacité aux semaines 18 et 52 d'une étude sur l'empagliflozine avec contrôle placebo comme traitement d'appoint à plusieurs doses d'insuline par jour avec ou sans metformine
Placebo Empagliflozine10 mg Empagliflozine25 mg
N 188 186 189
HbA1c (%) à la semaine 18
Valeur initiale (moyenne) 8,33 8,39 8,29
Variation par rapport à la valeur -0,50 -0,94 -1,02
initiale1
Différence par rapport au -0,44* (-0,61, -0,52* (-0,69,
placebo1 (IC 97,5 %) -0,27) -0,35)
N 115 119 118
HbA1c (%) à la semaine 522
Valeur initiale (moyenne) 8,25 8,40 8,37
Variation par rapport à la valeur -0,81 -1,18 -1,27
initiale1
Différence par rapport au -0,38*** (-0,62, -0,46* (-0,70,
placebo1 (IC 97,5 %) -0,13) -0,22)
N 113 118 118
Patients (%) atteignant une 26,5 39,8 45,8
valeur d'HbA1c <7 % avec une
valeur initiale d'HbA1c ≥7 % à la
semaine 52
N 115 118 117
Dose d'insuline (UI/jour) à la
semaine 522
Valeur initiale (moyenne) 89,94 88,57 90,38
Variation par rapport à la valeur 10,16 1,33 -1,06
initiale1
Différence par rapport au -8,83# (-15,69, -11,22** (-18,09,
placebo1 (IC 97,5 %) -1,97) -4,36)
N 115 119 118
Poids corporel (kg) à la semaine
522
Valeur initiale (moyenne) 96,34 96,47 95,37
Variation par rapport à la valeur 0,44 -1,95 -2,04
initiale1
Différence par rapport au -2,39* (-3,54, -2,48* (-3,63,
placebo1 (IC 97,5 %) -1,24) -1,33)
1 Moyenne ajustée par rapport à la valeur initiale
2 semaine 19-40: régime Treat-to-Target pour l'ajustement de la dose d'insuline, pour l'atteinte des valeurs cibles prédéfinies de la glycémie (préprandiale <100 mg/dl (5,5 mmol/l), postprandiale <140 mg/dl (7,8 mmol/l)
* valeur de p <0,0001
** valeur de p = 0,0003
*** valeur de p = 0,0005
# valeur de p = 0,0040
Empagliflozine deux fois par jour versus une fois par jour comme traitement d'appoint à la metformine
L'efficacité et la sécurité de l'empagliflozine administrée deux fois par jour versus une fois par jour (dose journalière de 10 mg et 25 mg) comme traitement d'appoint chez les patients dont la glycémie était insuffisamment contrôlée sous monotraitement par la metformine ont été évaluées au cours d'une étude en double aveugle, contrôlée contre placebo pendant 16 semaines. Tous les traitements avec l'empagliflozine ont induit une réduction significative de l'HbA1c entre l'inclusion (valeur moyenne globale 7,8 %) et après 16 semaines de traitement en comparaison avec le placebo. L'administration d'empagliflozine deux fois par jour a conduit à une réduction de l'HbA1c comparable à celle obtenue avec le schéma thérapeutique comportant une administration une fois par jour, la différence en termes de réduction de l'HbA1c entre l'inclusion et la semaine 16 est de -0,02 % (IC 95 % -0,16; 0,13) pour l'empagliflozine 5 mg deux fois par jour versus 10 mg une fois par jour et -0,11 % (IC 95 % -026; 0,03) pour l'empagliflozine 12,5 mg deux fois par jour versus 25 mg une fois par jour.
Evénements cardiovasculaires chez des patients présentant une affection cardiovasculaire manifeste
L'étude EMPA-REG a comparé le risque d'événements cardiovasculaires chez des patients présentant un diabète de type 2 et un antécédent cardiovasculaire sous traitement par empagliflozine versus placebo. Un total de 7020 patients présentant une coronaropathie (pathologie concernant un ou plusieurs vaisseaux), après un infarctus du myocarde (IM), après un accident vasculaire cérébral et/ou une maladie artérielle occlusive périphérique a été inclus dans l'étude. Ils ont été traités pendant une durée allant jusqu'à 4,5 ans (durée de traitement médiane 3,1 ans), en plus d'un traitement existant, soit par empagliflozine 10 mg (n=2.345), soit par empagliflozine 25 mg (n=2.342), soit par un placebo (n=2.333).
72,4 % de la population était caucasienne, 21,6 % asiatique et 5,1 % noire. L'âge moyen était de 63 ans (9,3 % des patients ≥75 ans), 71,5 % des patients étaient de sexe masculin. Au début de l'étude, environ 81 % des patients recevaient un inhibiteur du système rénine-angiotensine, 65 % des bêtabloquants, 43 % des diurétiques, 89 % des anticoagulants et 81 % une médication hypolipémiante. Au début de l'étude, environ 74 % des patients prenaient de la metformine, 48 % utilisaient de l'insuline et 43 % recevaient une sulfonylurée. Au cours des 12 premières semaines, le traitement a été observé sans autres modifications. A partir de cette date, le traitement (y compris hypertension et dyslipidémie) pouvait être modifié selon les directives thérapeutiques actuelles.
Le critère d'évaluation primaire (défini pour les deux bras empagliflozine ensemble) était la durée jusqu'au premier événement du critère d'évaluation combiné composé des décès cardiovasculaires, des IM non fatals et des accidents vasculaires cérébraux non fatals (3-point MACE [Major Adverse Cardiovascular Events]). Tous les décès cardiovasculaires ont été catégorisés par un comité d'experts en aveugle (CEC) comme IM fatal, accident vasculaire cérébral fatal, décès suite à une insuffisance cardiaque (par progression ou choc cardiogène), décès subit ou autres événements cardiovasculaires fatals.
Les analyses par sous-groupes pour 3P-MACE et décès cardiovasculaire après traitement antidiabétique au début de l'étude (dont metformine et insuline) affichaient des effets de traitement conformes à ceux de la population totale, dans le cadre d'une estimation des points pour le hazard ratio de la population totale s'inscrivant dans les intervalles de confiance des sous-groupes pour chaque critère d'évaluation.
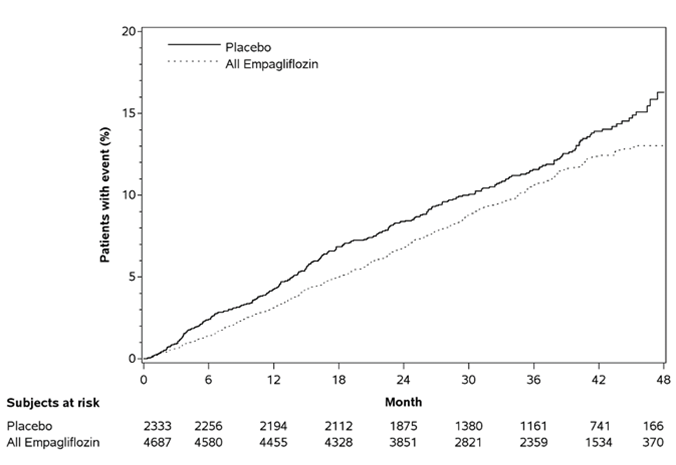
Réduction du risque de décès cardiovasculaires et mortalité totale
L'empagliflozine était supérieure au placebo en ce qui concerne le critère d'évaluation primaire (c'est-à-dire qu'elle provoquait une diminution du risque d'événements cardiovasculaires). Cet effet n'a pas pu être expliqué uniquement par la différence entre les groupes de traitement dans le contrôle de la glycémie et reposait principalement sur la réduction marquée de l'incidence de décès cardiovasculaires (Tableau 5 et Figure 1). Un effet préventif a été observé pour toutes les catégories de décès cardiovasculaires et était le plus net pour le décès pour cause d'insuffisance cardiaque progressive. Parallèlement, le risque d'infarctus du myocarde non fatals n'a pas été réduit de façon statistiquement significative. L'empagliflozine n'a également eu aucun effet préventif pour les accidents vasculaires cérébraux et a provoqué une augmentation numérique du risque (HR [intervalle de confiance 95 %] d'événements fatals/non fatals: 1,18 [0,89, 1,56]). En outre, il s'est produit une réduction du critère d'évaluation primaire chez les patients qui ont été inclus dans l'étude exclusivement en raison d'antécédents cérébrovasculaires. La plus forte diminution de la mortalité cardiovasculaire a été observée chez les patients présentant 2 ou 3 facteurs de risque.
En raison principalement de la réduction des décès cardiovasculaires (l'empagliflozine n'a pas provoqué de réduction statistiquement significative de la mortalité non cardiovasculaire), la mortalité totale s'est également améliorée dans le cadre du traitement par empagliflozine.
Tableau 5: Effet de traitement eu égard au critère d'évaluation primaire combiné, ses composants et la mortalité (population traitée*)
Placebo Empagliflozine(10
et 25 mg, en pool)
N 2333 4687
Durée jusqu'à la première occurrence d'un décès 282 (12,1) 490 (10,5)
cardiovasculaire, IM non fatal ou accident vasculaire
cérébral non fatal
Hazard ratio vs. placebo (IC 95,02 %)** 0,86 (0,74; 0,99)
Valeur p de supériorité 0,0382
Décès cardiovasculaires N (%) 137 (5,9) 172 (3,7)
Hazard ratio vs. placebo (IC 95 %) 0,62 (0,49; 0,77)
Valeur p <0,0001
IM non fatals N (%) 121 (5,2) 213 (4,5)
Hazard ratio vs. placebo (IC 95 %) 0,87 (0,70; 1,09)
Valeur p 0,2189
Accidents vasculaires cérébraux non fatals N (%) 60 (2,6) 150 (3,2)
Hazard ratio vs. placebo (IC 95 %) 1,24 (0,92; 1,67)
Valeur p 0,1638
Mortalité totale N (%) 194 (8,3) 269 (5,7)
Hazard ratio vs. placebo (IC 95 %) 0,68 (0,57; 0,82)
Valeur p <0,0001
Mortalité non cardiovasculaire N (%) 57 (2,4) 97 (2,1)
Hazard ratio vs. placebo (IC 95 %) 0,84 (0,60; 1,16)
* c'est-à-dire patients qui ont reçu au moins une dose du médicament à l'étude
** Puisque les données de l'étude ont été prises en compte dans une analyse intermédiaire, un intervalle de confiance bilatéral de 95,02 % s'applique, conforme à une valeur p inférieure à 0,0498 pour le caractère significatif
Figure 1: Durée jusqu'à la première occurrence d'un événement du critère d'évaluation primaire (décès cardiovasculaire/IM non fatal/accident vasculaire cérébral non fatal)
Dans une étude croisée randomisée avec contrôle placebo et comparateur actif portant sur 30 volontaires sains, aucune augmentation de l'intervalle QTc n'a été observée sous 25 mg ou 200 mg d'empagliflozine.
Glycémie postprandiale (à 2 heures)
Le traitement par empagliflozine en appoint à la metformine ou à la metformine et une sulfonylurée a entraîné une réduction cliniquement significative de la glycémie postprandiale à 2 heures (test de tolérance au repas) à 24 semaines (traitement d'appoint à la metformine: placebo +5,9 mg/dl [+0,33 mmol/l], empagliflozine 10 mg: -46,0 mg/dl [-2,56 mmol/l]). Traitement d'appoint à la metformine et à une sulfonylurée: placebo -2,3 mg/dl [-0,13 mmol/l], empagliflozine 10 mg: -35,7 mg/dl [1,98 mmol/l].
Patients présentant une valeur initiale d'HbA1c ≥9 %
Dans une analyse prédéfinie de patients avec une valeur initiale d'HbA1c ≥9,0 %, le traitement a entraîné à la semaine 24 une réduction de l'HbA1c statistiquement significative (variation moyenne corrigée par rapport à la valeur initiale de -1,40 % pour l'empagliflozine 10 mg et de -0,44 % pour le placebo).
Poids corporel
Dans une analyse poolée prédéfinie de 4 études contrôlées versus placebo, le traitement par empagliflozine (68 % de l'ensemble des patients recevaient de la metformine comme traitement de fond) a entraîné une réduction du poids corporel (-2,04 kg pour l'empagliflozine 10 mg, -0,24 kg pour le placebo) à la semaine 24, qui s'est maintenue jusqu'à la semaine 52 (-1,96 kg pour l'empagliflozine 10 mg, -0,16 kg pour le placebo).
Pression artérielle
L'efficacité et la tolérance de l'empagliflozine ont été évaluées au cours d'une étude en double aveugle avec contrôle placebo d'une durée de 12 semaines portant sur des patients avec diabète sucré de type 2 et une hypertension artérielle, traités par divers médicaments antidiabétiques et jusqu'à 2 médicaments antihypertenseurs. Le traitement par empagliflozine une fois par jour a entraîné une amélioration statistiquement significative de l'HbA1c et de la pression artérielle systolique et diastolique moyenne sur 24 heures, déterminée par un suivi ambulatoire de la pression artérielle (Tableau 6). Le traitement par empagliflozine a entraîné une réduction de la PAS et de la PAD en position assise.
Tableau 6: Résultats d'une étude avec contrôle placebo avec l'empagliflozine chez des patients avec diabète sucré de type 2 et pression artérielle non contrôlée (Full Analysis Set) après 12 semaines (LOCF)
Placebo Empagliflozine10 mg Empagliflozine25 mg
N 271 276 276
HbA1c (%) à la semaine 12
Valeur initiale (moyenne) 7,90 7,87 7,92
Variation par rapport à la valeur initiale1 0,03 -0,59 -0,62
Différence par rapport au placebo1(IC 95 %) -0,62* (-0,72, -0,52) -0,65* (-0,75, -0,55)
PAS sur 24 heures à la semaine 12
Valeur initiale (moyenne) 131,72 131,34 131,18
Variation par rapport à la valeur initiale1 0,48 -2,95 -3,68
Différence par rapport au placebo1(IC 95 %) -3,44* (-4,78, -2,09) -4,16* (-5,50, -2,83)
PAD sur 24 heures à la semaine 12
Valeur initiale (moyenne) 75,16 75,13 74,64
Variation par rapport à la valeur initiale1 0,32 -1,04 -1,40
Différence par rapport au placebo1(IC 95 %) -1,36** (-2,15, -0,56) -1,72* (-2,51, -0,93)
1 Moyenne ajustée par rapport à la valeur initiale
* p <0,0001
** p < 0,001
Dans une analyse poolée prédéfinie de quatre études contrôlées versus placebo, le traitement par empagliflozine a entraîné une réduction de la pression artérielle systolique (empagliflozine 10 mg: -3,9 mmHg) par rapport au placebo (-0,5 mmHg) et une réduction de la pression artérielle diastolique (empagliflozine 10 mg: -1,8 mmHg) par rapport au placebo (-0,5 mmHg) à la semaine 24; ces améliorations étaient maintenues jusqu'à la semaine 52.
Paramètres de laboratoire
Augmentation de l'hématocrite
Dans une analyse combinée de la sécurité de toutes les études avec de la metformine comme traitement de fond, les modifications moyennes de l'hématocrite par rapport aux valeurs initiales étaient de 3,6 % pour l'empagliflozine 10 mg et de 4,0 % pour l'empagliflozine 25 mg par rapport à 0 % pour le placebo. Dans l'étude EMPA-REG Outcome, les valeurs d'hématocrite étaient redescendues jusqu'aux valeurs initiales à la fin de la phase de suivi de 30 jours après l'arrêt du médicament.
Augmentation des lipides sériques
Dans une analyse combinée de la sécurité de toutes les études avec de la metformine comme traitement de fond, le pourcentage d'augmentation moyen par rapport aux valeurs initiales pour l'empagliflozine 10 mg ou 25 mg comparé au placebo était de 5,0 % ou 5,2 % versus 3,7 % pour le cholestérol total; de 4,6 % ou 2,7 % versus -0,5 % pour le cholestérol HDL; de 9,1 % ou 8,7 % versus 7,8 % pour le cholestérol LDL; de 5,4 % ou 10,8 % versus 12,1 % pour les triglycérides.
Enfants et adolescents
L'efficacité clinique et la sécurité de l'empagliflozine 10 mg une fois par jour (avec possibilité d'une augmentation de la dose à 25 mg une fois par jour) ou de la linagliptine 5 mg une fois par jour ont été étudiées chez des enfants et des adolescents de 10 à 17 ans atteints de diabète sucré de type 2 dans le cadre d'une étude (DINAMO) en groupes parallèles, contrôlée contre placebo, randomisée et menée en double aveugle sur une période de 26 semaines, avec une phase d'extension en double aveugle d'une durée allant jusqu'à 52 semaines visant à évaluer la sécurité du traitement actif. Au total, 157 patients ont reçu l'empagliflozine (10 mg ou 25 mg; N = 52), la linagliptine (N = 52) ou le placebo (N = 53). Parmi les traitements de fond administrés en complément de mesures diététiques et d'une activité physique figuraient la metformine en monothérapie (51%), la metformine associée à l'insuline (40,1%) et l'insuline (3,2%), ou aucun traitement de fond n'a été administré (5,7%). La valeur initiale d'HbA1c moyenne était de 8,03%. La population étudiée comprenait 38,2% de patients de sexe masculin et 61,8% de patients de sexe féminin, avec une moyenne d'âge de 14,5 ans (extrêmes: 10 à 17 ans); 51,6% avaient au moins 15 ans. La population étudiée était en outre composée comme suit: 49,7% des participants étaient d'origine caucasienne, 5,7% d'origine asiatique et 31,2% d'origine africaine/afro-américaine. L'IMC moyen s'élevait à 36,04 kg/m², le poids corporel moyen à 99,92 kg. Seuls les patients avec un DFGe ≥60 ml/min/1,73 m² ont été inclus dans l'étude DINAMO.
L'empagliflozine s'est avérée supérieure au placebo en ce qui concerne le critère d'évaluation principal, à savoir la réduction de l'HbA1c à 26 semaines par rapport à la valeur initiale, et ce, indépendamment d'un traitement de secours ou d'un arrêt du traitement. En outre, le traitement par l'empagliflozine a entraîné une réduction cliniquement significative de la GAJ (Tableau 7).
Tableau 7: Résultats d'une étude de l'empaglifozine contrôlée contre placebo, d'une durée de 26 semaines, menée chez des enfants et des adolescents atteints de diabète sucré de type 2 (analyse modifiée en intention de traiter)
Placebo Empagliflozine (10 et 25 mg une fois
par jour, données poolées)
N 53 52
HbA1c (%)¹
Valeur initiale (moyenne) 8,05 8,00
Variation par rapport à la valeur 0,68 -0,17
initiale²
Différence par rapport au placebo² -0,84 (-1,50, -0,19)
(IC à 95%)
Valeur p pour la supériorité 0,0116
N 52 48
GAJ (mg/dl) [mmol/l] 3,4
Valeur initiale (moyenne) 158,6 [8,80] 154,4 [8,57]
Variation par rapport à la valeur 15,7 [0,87] -19,5 [-1,08]
initiale²
Différence par rapport au placebo² -35,2 (-58,6, -11,7) [-1,95 (-3,25,
(IC à 95%) -0,65)]
Valeur p nominale 0,0035
¹ Imputation multiple avec 500 itérations pour données manquantes
² Moyenne ajustée en fonction de la valeur initiale et de la stratification
³ Last Observation Carried Forward (LOCF), y compris les valeurs initiales
4 Non évaluée sur le plan de la signification statistique; non incluse dans la procédure de test séquentiel
PharmacocinétiqueJardiance Met
Les études de bioéquivalence menées auprès de sujets sains montrent que les comprimés de l'association Jardiance Met (empagliflozine/chlorhydrate de metformine) dosés à 5 mg/500 mg, 5 mg/850 mg et 5 mg/1000 mg sont bioéquivalents à l'administration concomitante d'empagliflozine et de metformine en comprimés distincts aux dosages correspondants.
Les données qui suivent proviennent d'études qui ont été réalisées soit avec l'empagliflozine soit avec la metformine.
Empagliflozine
Absorption
La pharmacocinétique de l'empagliflozine a été largement étudiée chez les volontaires sains et les patients présentant un diabète de type 2. Après administration par voie orale, l'empagliflozine a été rapidement absorbée, avec des concentrations plasmatiques maximales survenant 1,5 heures (tmax médian) après la prise de la dose. Ensuite, les concentrations plasmatiques ont diminué de manière biphasique avec une phase de distribution rapide et une phase terminale relativement lente. Pour une administration une fois par jour (qd) de 10 mg d'empagliflozine, l'ASC plasmatique moyenne à l'état d'équilibre était de 1870 nmol.h/l et la Cmax 259 nmol/l. L'exposition systémique à l'empagliflozine a augmenté proportionnellement à la dose (domaine de 10 mg-100 mg). La pharmacocinétique de l'empagliflozine est linéaire par rapport au temps. La pharmacocinétique de l'empagliflozine n'a pas montré de différence cliniquement significative entre les volontaires sains et les patients avec un diabète de type 2.
La pharmacocinétique d'une dose de 5 mg administrée deux fois par jour d'empagliflozine et d'une dose de 10 mg d'empagliflozine administrée une fois par jour a été comparée chez les sujets sains. L'exposition globale (ASC) à l'empagliflozine sur 24 heures pour une administration de 5 mg deux fois par jour et pour une administration de 10 mg une fois par jour était similaire. De manière prévisible, l'administration de 5 mg d'empagliflozine deux fois par jour en comparaison avec l'administration de 10 mg d'empagliflozine une fois par jour a entraîné une Cmax plus faible et des concentrations plasmatiques minimales (Cmin) d'empagliflozine plus élevées.
L'administration d'empagliflozine 25 mg après la prise d'un repas à forte teneur en graisse et en calories a entraîné une faible baisse de la biodisponibilité; l'ASC a diminué d'environ 16 % et la Cmax d'environ 37 % par rapport à une prise à jeun. L'effet observé des aliments sur la pharmacocinétique de l'empagliflozine n'a pas été jugé cliniquement significatif et l'empagliflozine peut être administrée au cours ou en dehors des repas.
Distribution
D'après l'analyse pharmacocinétique de population, le volume de distribution apparent à l'état d'équilibre a été estimé à 73,8 l. Après l'administration orale d'une solution d'empagliflozine marquée au [14C] à des volontaires sains, la répartition érythrocytaire était de 36,8 % environ et la liaison aux protéines plasmatiques de 86,2 %.
Métabolisme
Dans une étude avec de l'empagliflozine marqué au [14C], la plus grande partie de la radioactivité totale dans le plasma a pu être attribuée à l'empagliflozine inchangée.
Les métabolites les plus fréquents étaient trois glucuronides conjugués (2-O-, 3-O- et 6-Oglucuronides). L'exposition systémique à chaque métabolite était inférieure à 10 % de la radioactivité totale dans le plasma. Les données in vitro suggèrent que la voie primaire du métabolisme de l'empagliflozine chez l'homme est la glucuronidation par les uridine-5'-diphosphoglucuronosyltransférases UGT1A3, UGT1A8, UGT1A9 et UGT2B7.
Élimination
D'après l'analyse pharmacocinétique de population, la demi-vie d'élimination terminale apparente de l'empagliflozine a été estimée à 12,4 heures et la clairance orale apparente à 10,6 l/heure. Les variabilités interindividuelles et résiduelles pour la clairance orale de l'empagliflozine étaient de 39,1 %. Avec une administration une fois par jour, les concentrations plasmatiques d'empagliflozine à l'état d'équilibre étaient atteintes à la cinquième dose. En cohérence avec la demi-vie, une accumulation allant jusqu'à 22 % a été observée à l'état d'équilibre pour l'ASC plasmatique. Après l'administration orale d'une solution d'empagliflozine marquée au [14C] à des volontaires sains, environ 41,2 % de la radioactivité était éliminée dans les fèces et 54,4 % dans l'urine. La substance mère sous forme inchangée formait la majorité de la radioactivité retrouvée dans les fèces (82,9 %) et 43,5 % de la radioactivité liée au principe actif excrétée dans l'urine.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Chez les sujets présentant une insuffisance hépatique légère, modérée ou sévère selon la classification de Child-Pugh, l'ASC de l'empagliflozine a augmenté d'environ 23 %, 47 % et 75 % et la Cmax d'environ 4 %, 23 % et 48 % respectivement, par rapport aux sujets avec une fonction hépatique normale. Sur la base des données pharmacocinétiques, un ajustement de la dose n'est donc pas nécessaire chez les patients insuffisants hépatiques.
Troubles de la fonction rénale
Chez les patients avec une insuffisance rénale légère (DFGe: 60 - <90 ml/min/1,73 m2), moyenne (DFGe: 30 - <60 ml/min/1,73 m2) et sévère (DFGe: <30 ml/min/1,73 m2) et les patients présentant une insuffisance rénale terminale, l'ASC de l'empagliflozine a augmenté d'environ 18 %, 20 %, 66 % et 48 % respectivement, par rapport aux patients avec une fonction rénale normale. Les concentrations plasmatiques maximales d'empagliflozine étaient similaires chez les patients présentant une insuffisance rénale modérée et une insuffisance rénale terminale par rapport aux patients ayant une fonction rénale normale. Les concentrations plasmatiques maximales d'empagliflozine étaient environ 20 % plus élevées chez les patients présentant une insuffisance rénale légère et modérée par rapport aux patients ayant une fonction rénale normale. En cohérence avec l'étude de phase I, l'analyse pharmacocinétique de population a montré que la baisse du DFGe allait de pair avec une réduction apparente de la clairance orale de l'empagliflozine et entraînait ainsi une augmentation de l'exposition systémique. Sur la base des données pharmacocinétiques, un ajustement de la dose n'est donc pas nécessaire chez les patients insuffisants rénaux.
Indice de masse corporelle (IMC)
Un ajustement de la dose sur la base de l'IMC n'est pas nécessaire. Selon l'analyse pharmacocinétique de population, l'indice de masse corporelle n'a aucun effet cliniquement significatif sur la pharmacocinétique de l'empagliflozine.
Sexe
Un ajustement de la dose sur la base du sexe n'est pas nécessaire. Selon l'analyse pharmacocinétique de population, le sexe n'a aucun effet cliniquement significatif sur la pharmacocinétique de l'empagliflozine.
Origine ethnique
Selon l'analyse pharmacocinétique de population, l'estimation de l'ASC était 13,5 % plus élevée chez les sujets asiatiques avec un IMC de 25 kg/m2 que chez les sujets non asiatiques avec un IMC de 25 kg/m2.
Patients âgés
Selon l'analyse pharmacocinétique de population, l'âge n'a aucun effet cliniquement significatif sur la pharmacocinétique de l'empagliflozine.
Enfants et adolescents
La pharmacocinétique et la pharmacodynamique d'une dose unique d'empagliflozine 5 mg, 10 mg et 25 mg ont été étudiées chez des enfants et des adolescents âgés de 10 à 17 ans atteints de diabète sucré de type 2. La pharmacocinétique observée et la relation pharmacocinétique/pharmacodynamique (élimination urinaire du glucose) chez les patients adultes et pédiatriques ont été comparables après la prise en compte des covariables significatives.
La pharmacocinétique et la pharmacodynamique (variation de l'HbA1c par rapport à la valeur initiale) de l'empagliflozine 10 mg avec possibilité d'une augmentation à 25 mg ont été étudiées chez des enfants et des adolescents de 10 à 17 ans atteints de diabète sucré de type 2. La relation exposition-effet observée a été généralement comparable chez les adultes, les enfants et les adolescents. L'administration orale d'empagliflozine a entraîné une exposition dans la plage observée chez les patients adultes. Les moyennes géométriques observées pour les concentrations résiduelles et les concentrations à 1,5 heure post-administration à l'état d'équilibre ont été de 26,6 nmol/l et 308 nmol/l pour l'empagliflozine 10 mg une fois par jour, et de 67,0 nmol/l et 525 nmol/l pour l'empagliflozine 25 mg une fois par jour.
Metformine
Absorption
Après administration orale de metformine, le Tmax est atteint après 2,5 h. La biodisponibilité absolue d'une dose de 500 mg ou de 850 mg de chlorhydrate de metformine en comprimés est d'environ 50 % à 60 % chez le sujet sain. Après administration d'une dose orale, la fraction non résorbée qui a été retrouvée dans les fèces était de 20 à 30 %.
Après administration orale d'une dose unique de 500 à 2500 mg, on a observé une hausse proportionnellement inférieure de la Cmax en relation probablement avec un mécanisme saturable. Aux posologies usuelles de metformine, les concentrations plasmatiques à l'état d'équilibre sont atteintes en 24 à 48 heures et sont généralement inférieures à 1 μg/ml. Dans les essais cliniques contrôlés, les concentrations plasmatiques maximales Cmax observées n'ont pas excédé 4 μg/ml, même aux doses maximales.
La nourriture diminue et ralentit l'absorption de la metformine. Après administration d'une dose de 850 mg et la prise d'aliments, on a noté une diminution de la Cmax de 40 %, une diminution de 25 % de l'ASC, un Tmax prolongé de 35 minutes. La signification clinique de ces modifications reste inconnue.
Distribution
La liaison aux protéines plasmatiques de la metformine est négligeable. La metformine diffuse en partie dans les érythrocytes. Le pic sanguin est plus faible que le pic plasmatique et apparaît approximativement au même moment. Les érythrocytes représentent probablement un compartiment secondaire de distribution. Le volume de distribution moyen se situe à 63–276 l.
On ignore si la metformine passe la barrière placentaire et si elle parvient dans le lait maternel.
Chez la rate, de faibles quantités sont retrouvées dans le lait.
Métabolisme
La metformine n'est pas métabolisée chez l'être humain.
Élimination
La metformine est excrétée dans l'urine sous forme inchangée. La clairance rénale est >400 ml/min et donc environ 3,5 fois plus élevée que la clairance de la créatinine. L'élimination intervient donc principalement par sécrétion tubulaire active. Après administration orale, la demi-vie d'élimination plasmatique est d'environ 6,5 heures. Mesurée dans le sang complet, la demi-vie se situe à environ 17,6 heures.
En cas de fonction rénale normale, la metformine ne s'accumule pas dans l'organisme à la dose usuelle recommandée (1500-2000 mg).
Cinétique pour certains groupes de patients
Troubles de la fonction rénale
En cas d'atteinte de la fonction rénale, la clairance rénale est diminuée proportionnellement à celle de la clairance de la créatinine ou au DFG, c'est-à-dire que la demi-vie d'élimination est prolongée avec un risque d'accumulation.
Données précliniquesEmpagliflozine et metformine
Dans le cadre d'une étude évaluant la toxicité globale pendant une durée de 13 semaines chez le rat comportant l'administration de l'empagliflozine associée à la metformine, le NOAEL (No Observed Adverse Effect Level) a été observé à des doses de 50/100 mg/kg/jour d'empagliflozine/de metformine sur la base de l'hypochlorémie, c'est-à-dire après une exposition qui correspondait environ à 4 l'exposition clinique pour l'empagliflozine et à 2,1 l'exposition clinique pour la metformine sur la base de l'ASC à une dose journalière de 25 mg d'empagliflozine et de 2000 mg de metformine.
Toxicité sur la reproduction
Dans le cadre d'une étude portant sur le développement embryonnaire et fœtal réalisée chez des rates gestantes [(Wistar resp. Crl:WI (Han)] comportant une administration correspondant à 14 fois l'exposition clinique sur la base de l'ASC pour une dose de 25 mg d'empagliflozine et à 4 fois l'exposition clinique sur la base de l'ASC pour une dose de 2000 mg de metformine, aucun effet tératogène qui serait dû à l'utilisation concomitante d'empagliflozine et de metformine n'a été mis en évidence. À des doses de 300/600 mg/kg/jour d'empagliflozine/metformine en association ou à une dose de 600 mg/kg/jour de metformine en monothérapie correspondant à 8 fois l'exposition clinique sur la base de l'ASC à une dose de 2000 mg de metformine, la metformine s'est avérée tératogène.
Les données qui suivent proviennent d'études qui ont été réalisées soit avec l'empagliflozine soit avec la metformine.
Empagliflozine
Dans les études de toxicité à long terme sur les rongeurs et les chiens, des signes de toxicité ont été observés à des expositions supérieures ou égales à 10 fois la dose de 25 mg. La toxicité était généralement cohérente avec la pharmacologie secondaire liée à l'excrétion urinaire de glucose, notamment la perte de poids et de graisse corporelle, l'augmentation de la consommation alimentaire, la diarrhée, la déshydratation, la baisse du glucose sérique et les augmentations d'autres paramètres sériques reflétant une augmentation du métabolisme protéique, une gluconéogenèse et un déséquilibre électrolytique, des modifications urinaires (polyurie et glycosurie) et des modifications microscopiques des reins (dont une dilatation tubulaire et une minéralisation tubulaire et pelvienne).
Carcinogénicité
L'empagliflozine n'a pas augmenté l'incidence de tumeurs chez des rats femelles jusqu'à la dose maximale de 700 mg/kg/jour (correspondant à environ 72 fois l'exposition clinique sur la base de l'ASC pour une dose de 25 mg). Chez les rats mâles, des lésions prolifératives vasculaires bénignes (hémangiomes) du ganglion lymphatique mésentérique, liées au traitement, ont été observées à la dose de 700 mg/kg/jour (correspondant à environ 42 fois l'exposition clinique pour une dose de 25 mg). Ces tumeurs sont fréquentes chez le rat et ne sont vraisemblablement pas pertinentes pour l'homme.
L'empagliflozine n'a pas augmenté l'incidence de tumeurs chez des souris femelles jusqu'à la dose maximale de 1000 mg/kg/jour (correspondant à environ 62 fois l'exposition clinique sur la base de l'ASC pour une dose de 25 mg). Chez des souris mâles, l'empagliflozine a induit une augmentation des tumeurs rénales à la dose de 1000 mg/kg/jour (correspondant à environ 45 fois l'exposition clinique pour une dose de 25 mg). Le mécanisme d'action entraînant l'apparition de ces tumeurs, est dépendant de la prédisposition naturelle de la souris CD-1 mâle aux pathologies rénales et à une voie métabolique non superposable à l'homme. C'est pourquoi les tumeurs rénales des souris CD-1 mâles sont considérées comme vraisemblablement non pertinentes pour l'homme.
Génotoxicité
L'empagliflozine n'est pas génotoxique.
Toxicité sur la reproduction
L'empagliflozine administrée à des rates [(Wistar resp. Crl:WI (Han)] et des lapines pendant l'organogenèse à des doses jusqu'à 300 mg/kg (correspondant à environ 48 fois et 128 l'exposition clinique sur la base de l'ASC pour une dose de 25 mg) n'était pas tératogène. L'exposition à des doses d'empagliflozine toxiques pour la mère et correspondant à environ 155 fois l'exposition clinique pour une dose de 25 mg, a également causé une incurvation des os des membres chez le rat. L'exposition à des doses d'empagliflozine toxiques pour la mère et correspondant à environ 139 fois l'exposition clinique pour une dose de 25 mg, a également causé une augmentation des pertes embryo-fœtales chez le lapin.
Dans les études de toxicité pré- et post-natales chez le rat, une diminution du gain de poids de la progéniture a été observée à des expositions maternelles environ 4 fois supérieures à l'exposition clinique pour une dose de 25 mg.
Dans une étude de toxicité chez de jeunes rats, lors de l'administration d'empagliflozine du jour 21 au jour 90 chez les femelles et les mâles, une minéralisation rénale tubulaire minimale non toxique mais dose-dépendante et non réversible a été observée après la naissance à partir d'une dose de 10 mg/kg/jour (correspondant environ à 0,6 fois l'exposition clinique pour une dose de 25 mg). Une dilatation tubulaire rénale et pelvienne minimale à légère a été observée uniquement à des doses de 100 mg/kg/jour (correspondant environ à 11 fois la dose clinique pour une dose 25 mg) qui n'était plus détectable après une phase de régénération de 13 semaines sans administration médicamenteuse.
Metformine
Des données issues d'études précliniques, basées sur des essais conventionnels portant sur la sécurité pharmacologique, la toxicité après administration répétée, la génotoxicité (test d'Ames, test de mutagénicité, test d'aberration chromosomique, test du micronoyau) et la carcinogénicité (chez les rongeurs à des doses allant jusqu'à 900 mg/kg/jour (rat) et 1500 mg/kg/jour (souris)) n'ont fourni aucun indice suggérant des risques particuliers pour l'application chez l'homme.
Toxicité sur la reproduction
La metformine n'a aucun effet sur la fertilité.
Toutefois, la metformine était tératogène chez les rats en association avec l'empagliflozine à une dose de 300/600 mg/kg/jour d'empagliflozine/metformine et en monothérapie à partir d'une dose de 500 mg/kg/jour [(Wistar resp. Crl:WI (Han)] (voir ci-dessus "Empagliflozine et metformine" )
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention "EXP" sur l'emballage.
Remarques particulières concernant le stockage
Tenir hors de la portée et de la vue des enfants. Conserver à température ambiante (15-25°C).
Numéro d’autorisation65570 (Swissmedic)
PrésentationComprimés pelliculés à 5/500 mg: 60 et 180 [2x90] (B)
Comprimés pelliculés à 5/850 mg: 60 et 180 [2x90] (B)
Comprimés pelliculés à 5/1000 mg: 60 et 180 [2x90] (B)
Comprimés pelliculés à 12,5/500 mg: 60 et 180 [2x90] (B)
Comprimés pelliculés à 12,5/850 mg: 60 et 180 [2x90] (B)
Comprimés pelliculés à 12,5/1000 mg: 60 et 180 [2x90] (B)
Titulaire de l’autorisationBoehringer Ingelheim (Suisse) GmbH, Bâle.
Mise à jour de l’informationDécembre 2025
|