|
| Résultat de recherche - Ipro sur Lutathera 370 MBq/ml solution pour perfusion / Lutathera CA 370 MBq/ml solution pour perfusion |
| Information professionnelle sur Lutathera 370 MBq/ml solution pour perfusion / Lutathera CA 370 MBq/ml solution pour perfusion: | Novartis Pharma Schweiz AG |
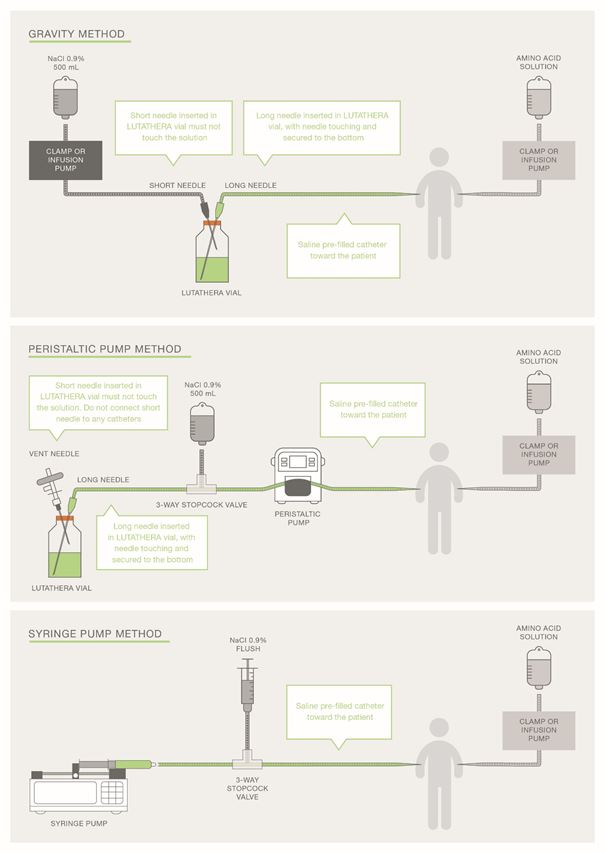
Posologie/Mode d’emploiLe médicament est réservé à l'usage hospitalier et ne peut être administré que par des médecins spécialisés titulaires du titre de formation postgraduée en médecine nucléaire. Composition Quantité
L-Lysine.HCl 25 g (équivalent à 20 g
de lysine)
L-Arginine.HCl 25 g (équivalent à 20,7
g d'arginine)
Solution de chlorure de sodium à 9 mg/ml (0,9%) pour préparations 1 l
injectables ou eau pour préparations injectables
Propriété Spécification L-Lysine.HCl Entre 18 et 25 g (équivalent à 14,4 à 20 g de L-lysine) L-Arginine.HCI Entre 18 et 25 g (équivalent à 14,9 à 20,7 g de L-arginine) Volume 1 l à 2 l Osmolalité < 1200 mOsmol/kg EI Sévérité des EI Ajustement posologique
Thrombocytopénie Première apparition de: Grade Suspendre la dose jusqu'à obtenir une
2 (nombre de plaquettes < 75 résolution complète ou partielle (grade 0
à 50 × 109/l) Grade 3 (nombre à 1). Reprendre Lutathera/Lutathera CA à
de plaquettes < 50 à 25 × la dose de 3700 MBq (100 mCi) pour les
109/l) Grade 4 (nombre de patients ayant obtenu une résolution
plaquettes < 25 × 109/l) complète ou partielle. Si une dose réduite
n'entraîne pas de thrombocytopénie de
grade 2, 3 ou 4, administrer
Lutathera/Lutathera CA à la dose de 7400
MBq (200 mCi) lors de l'administration
suivante. Arrêter définitivement
Lutathera/Lutathera CA en cas de
thrombocytopénie de grade ≥2 nécessitant
un intervalle entre les doses de plus de
16 semaines.
Grade 2, 3 ou 4 Arrêter définitivement le
récidivant traitement par Lutathera/Lutat
hera CA
Anémie et neutropénie Première apparition d'une Suspendre la dose jusqu'à obtenir une
anémie: Grade 3 (Hb < 8,0 résolution complète ou partielle (grade 0,
g/dl); transfusion indiquée 1 ou 2). Reprendre Lutathera/Lutathera CA
Grade 4 (conséquences à la dose de 3700 MBq (100 mCi) pour les
engageant le pronostic vital) patients ayant obtenu une résolution
Première apparition d'une complète ou partielle. Si une dose réduite
neutropénie: Grade 3 (nombre n'entraîne pas d'anémie ou de neutropénie
absolu de neutrophiles (NAN) de grade 3 ou 4, administrer
< 1,0 à 0,5 × 109/l) Grade 4 Lutathera/Lutathera CA à la dose de 7400
(NAN < 0,5 × 109/l) MBq (200 mCi) lors de l'administration
suivante. Arrêter définitivement
Lutathera/Lutathera CA en cas d'anémie ou
de neutropénie de grade ≥3 nécessitant un
intervalle entre les doses de plus de 16
semaines.
Grade 3 ou 4 récidivan Arrêter définitivement le
t traitement par Lutathera/Lutat
hera CA.
Toxicité rénale Première apparition de: Suspendre la dose jusqu'à obtenir une
-clairance de la créatinine résolution ou un retour aux valeurs
inférieure à 40 ml/min, initiales. Reprendre Lutathera/Lutathera
calculée en utilisant la CA à la dose de 3700 MBq (100 mCi) pour
formule de Cockcroft-Gault et les patients ayant obtenu une résolution
le poids corporel réel, ou ou un retour aux valeurs initiales. Si une
-augmentation de 40% de la dose réduite n'entraîne pas de toxicité
valeur initiale de la rénale, administrer Lutathera/Lutathera CA
créatinine sérique, ou à la dose de 7400 MBq (200 mCi) lors de
diminution de 40% de la l'administration suivante. Arrêter
clairance de la créatinine définitivement Lutathera/Lutathera CA en
par rapport à la valeur cas de toxicité rénale nécessitant un
initiale, calculée en intervalle entre les doses de plus de 16
utilisant la formule de semaines.
Cockcroft-Gault et le poids
corporel réel.
Toxicité rénale Arrêter définitivement
récidivante Lutathera/Lutathera CA
Hépatotoxicité Définie comme: -bilirubinémie Suspendre la dose jusqu'à obtenir une
> 3 fois la limite supérieure résolution ou un retour aux valeurs
de la normale (grade 3 ou 4), initiales. Reprendre Lutathera/Lutathera
ou albuminémie < 30 g/l avec CA à la dose de 3700 MBq (100 mCi) pour
INR > 1,5 les patients ayant obtenu une résolution
complète ou un retour aux valeurs
initiales. Si une dose réduite n'entraîne
pas d'hépatotoxicité, administrer
Lutathera/Lutathera CA à la dose de 7400
MBq (200 mCi) lors de l'administration
suivante. Arrêter définitivement
Lutathera/Lutathera CA en cas
d'hépatotoxicité nécessitant un intervalle
entre les doses de plus de 16 semaines.
Hépatotoxicité Arrêter définitivement
récidivante Lutathera/Lutathera CA
Tout autre EI de Première apparition d'un Suspendre la dose jusqu'à obtenir une
grade 3 ou de grade 4 grade 3 ou 4 résolution complète ou partielle (grade 0
selon CTCAE* à 2). Reprendre Lutathera/Lutathera CA à
la dose de 3700 MBq (100 mCi) pour les
patients ayant obtenu une résolution
complète ou partielle. Si une dose réduite
n'entraîne pas de toxicité de grade 3 ou
4, administrer Lutathera/Lutathera CA à la
dose de 7400 MBq (200 mCi) lors de
l'administration suivante. Arrêter
définitivement Lutathera/Lutathera CA en
cas d'EI de grade ≥3 nécessitant un
intervalle entre les doses de plus de 16
semaines.
Toxicité de grade 3 Arrêter définitivement
ou 4 récidivante Lutathera/Lutathera CA.
Aucun ajustement
posologique n'est
nécessaire en cas de
toxicités hématologiqu
es de grade 3 ou 4
qui sont uniquement
dues à une lymphopénie
. *CTCAE: Common
Terminology Criteria
for Adverse Events
(Critères communs de
terminologie pour les
événements indésirable
s), National Cancer
Institute
TMD: toxicité modifiant la dose Agents administrés Heure de début (min) Débit de perfusion(m Durée
l/h)
Antiémétiques délai de temps selon les indication selon les indication
suffisant avant la s de l'information s de l'information
solution d'acides professionnelle professionnelle
aminés
Solution d'acides aminés: soit la 0 250–500 en fonction 4 heures
préparation magistrale (1 l) soit du volume
la solution commerciale (1 à 2 l).
Lutathera/Lutathera CA avec 30 Jusqu'à 400 30 ± 10 minutes
solution de chlorure de sodium 9
mg/ml (0,9%) pour préparation
injectable
EXPOSITION AUX RAYONNEMENTS Organe Dose absorbée par l'organe (mGy/MBq) (n = 20)
Moyenne MS
Glandes surrénales 0,037 0,016
Cerveau 0,027 0,016
Seins 0,027 0,015
Paroi de la vésicule biliaire 0,042 0,019
Paroi du gros intestin inférieur 0,029 0,016
Intestin grêle 0,031 0,015
Paroi de l'estomac 0,031 0,015
Paroi du gros intestin supérieur 0,032 0,015
Paroi du cœur 0,032 0,015
Reins 0,654 0,295
Foie* 0,199 0,226
Poumons 0,031 0,015
Muscle 0,029 0,015
Ovaires** 0,031 0,013
Pancréas 0,038 0,016
Moelle osseuse rouge*** 0,035 0,029
Cellules ostéogéniques 0,151 0,268
Peau 0,027 0,015
Rate 0,846 0,804
Testicules**** 0,026 0,018
Thymus 0,028 0,015
Thyroïde 0,027 0,016
Paroi de la vessie 0,437 0,176
Utérus** 0,032 0,013
Organisme entier 0,052 0,027
Organe Dose de radiation absorbée par l'organe (mGy/MBq) (n = 10)
Valeur moyenne ET
Glandes surrénales 0,045 0,011
Cerveau 0,020 0,006
Seinsa 0,018 0,005
Paroi de la vésicule biliaire 0,030 0,010
Intestin grêle 0,045 0,011
Paroi de l'estomac 0,026 0,006
Paroi du cœur 0,023 0,006
Reins 0,778 0,280
Foie 0,210 0,205
Poumons 0,023 0,006
Ovairesb 0,026 0,007
Pancréas 0,027 0,006
Moelle osseuse rouge (sang)c 0,026 0,005
Moelle osseuse rouge (imagerie)c 0,057 0,027
Cellules ostéogènes 0,045 0,017
Rate 0,742 0,275
Testiculesd 0,020 0,005
Thymus 0,022 0,006
Thyroïde 0,027 0,017
Paroi de la vessie 0,552 0,089
Utérusb 0,030 0,008
Organisme entier 0,040 0,010
Hypophysee 1,114 0,425
Rectum 0,277 0,076
Côlon gauche 0,273 0,074
Côlon droit 0,156 0,041
Glandes salivaires 0,033 0,017
Prostated 0,025 0,006
Œsophage 0,023 0,006
Yeux 0,020 0,006
| |

| ||||||||||||
|
|